Mass spectrometry-based characterization of histone post-translational modification
IF 6.1
2区 生物学
Q1 BIOCHEMISTRY & MOLECULAR BIOLOGY
引用次数: 0
Abstract
Histone post-translational modifications (PTMs) play critical roles in regulating chromatin dynamics and gene expression. Increasing evidence demonstrates that the dysregulation of histone PTMs is closely associated with the pathogenesis of various diseases. Traditional methods for detecting histone PTMs, such as western blot (WB) and chromatin immunoprecipitation sequencing (ChIP-seq), are often limited by their dependence on specific antibodies and relatively low analytical throughput. Mass spectrometry (MS)-based proteomics offers a powerful and unbiased approach for comprehensive characterization of histone PTMs. This review focuses on the advanced development of MS-based strategies for characterizing histone PTMs. These strategies include histone extraction, enzymatic digestion, labeling, enrichment, and MS-based detection. These techniques not only enable comprehensive identification and quantitative analysis of classical modifications, such as acetylation and methylation, but also substantially facilitate the discovery of less-characterized histone PTMs, including succinylation, lactylation, crotonylation, and monoaminylation. Consequently, these findings significantly enhance the complexity of histone code. Collectively, MS-based approaches have profoundly advanced our understanding of histone PTM landscapes and their potential epigenetic regulatory mechanisms in both physiology and pathology contexts.
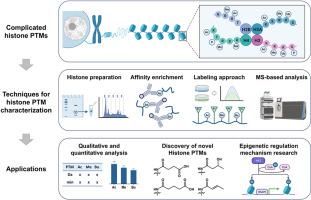
基于质谱的组蛋白翻译后修饰表征
组蛋白翻译后修饰(PTMs)在调节染色质动力学和基因表达中起着至关重要的作用。越来越多的证据表明,组蛋白ptm的失调与多种疾病的发病密切相关。检测组蛋白ptm的传统方法,如western blot (WB)和染色质免疫沉淀测序(ChIP-seq),往往受其依赖于特异性抗体和相对较低的分析通量的限制。基于质谱(MS)的蛋白质组学为组蛋白ptm的全面表征提供了一种强大而公正的方法。本文综述了基于ms的组蛋白ptm表征策略的最新进展。这些策略包括组蛋白提取、酶消化、标记、富集和质谱检测。这些技术不仅能够对经典修饰(如乙酰化和甲基化)进行全面的鉴定和定量分析,而且还极大地促进了发现较少表征的组蛋白PTMs,包括琥珀酰化、乳酸化、巴豆酰化和单胺化。因此,这些发现显著提高了组蛋白编码的复杂性。总的来说,基于ms的方法深刻地推进了我们对组蛋白PTM景观及其在生理和病理背景下潜在的表观遗传调控机制的理解。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Current Opinion in Chemical Biology
生物-生化与分子生物学
CiteScore
13.30
自引率
1.30%
发文量
113
审稿时长
74 days
期刊介绍:
COCHBI (Current Opinion in Chemical Biology) is a systematic review journal designed to offer specialists a unique and educational platform. Its goal is to help professionals stay informed about the growing volume of information in the field of Chemical Biology through systematic reviews.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


