Wudmir Y Rojas, Zargham Ahmad, Julia Jakiela, Helge Hecht, Jana Klánová, Elliott J Price
{"title":"Galaxy QCxMS for straightforward semi-empirical quantum mechanical EI-MS prediction.","authors":"Wudmir Y Rojas, Zargham Ahmad, Julia Jakiela, Helge Hecht, Jana Klánová, Elliott J Price","doi":"10.46471/gigabyte.160","DOIUrl":null,"url":null,"abstract":"<p><p>High-performance computing (HPC) environments are crucial for computational research, including quantum chemistry (QC), but pose challenges for non-expert users. Researchers with limited computational knowledge struggle to utilise domain-specific software and access mass spectra prediction for <i>in silico</i> annotation. Here, we provide a robust workflow that leverages interoperable file formats for molecular structures to ensure integration across various QC tools. The quantum chemistry package for mass spectral predictions after electron ionization or collision-induced dissociation has been integrated into the Galaxy platform, enabling automated analysis of fragmentation mechanisms. The extended tight binding quantum chemistry package, chosen for its balance between accuracy and computational efficiency, provides molecular geometry optimisation. A Docker image encapsulates the necessary software stack. We demonstrated the workflow for four molecules, highlighting the scalability and efficiency of our solution via runtime performance analysis. This work shows how non-HPC users can make these predictions effortlessly, using advanced computational tools without needing in-depth expertise.</p>","PeriodicalId":73157,"journal":{"name":"GigaByte (Hong Kong, China)","volume":"2025 ","pages":"gigabyte160"},"PeriodicalIF":1.2000,"publicationDate":"2025-07-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12257954/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"GigaByte (Hong Kong, China)","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.46471/gigabyte.160","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
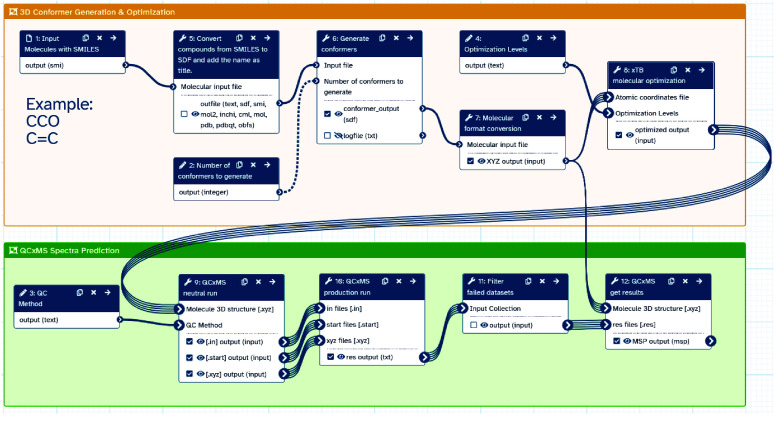
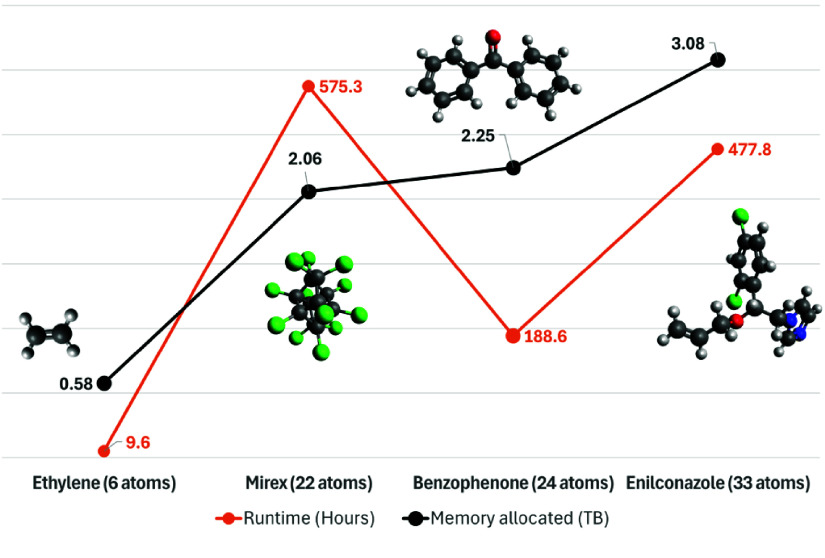
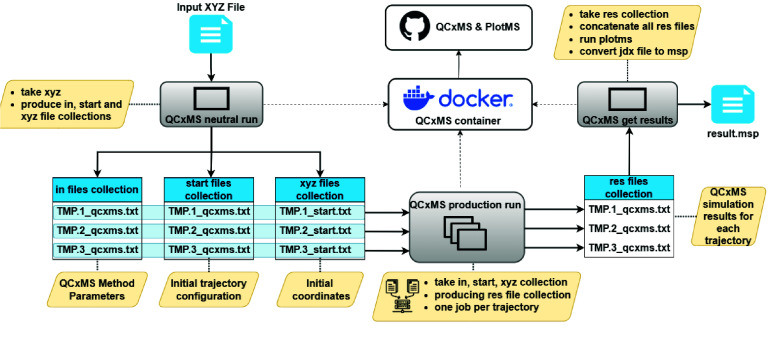
High-performance computing (HPC) environments are crucial for computational research, including quantum chemistry (QC), but pose challenges for non-expert users. Researchers with limited computational knowledge struggle to utilise domain-specific software and access mass spectra prediction for in silico annotation. Here, we provide a robust workflow that leverages interoperable file formats for molecular structures to ensure integration across various QC tools. The quantum chemistry package for mass spectral predictions after electron ionization or collision-induced dissociation has been integrated into the Galaxy platform, enabling automated analysis of fragmentation mechanisms. The extended tight binding quantum chemistry package, chosen for its balance between accuracy and computational efficiency, provides molecular geometry optimisation. A Docker image encapsulates the necessary software stack. We demonstrated the workflow for four molecules, highlighting the scalability and efficiency of our solution via runtime performance analysis. This work shows how non-HPC users can make these predictions effortlessly, using advanced computational tools without needing in-depth expertise.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


