{"title":"A molecular simulation-based deep neural network model for deciphering the adsorption of 5-Fluorouracil in COFs†","authors":"Khushboo Yadava and Ashutosh Yadav","doi":"10.1039/D5PM00007F","DOIUrl":null,"url":null,"abstract":"<p >A database of 1242 experimentally synthesized COFs has been studied to understand their potential as drug carriers by employing molecular simulations and machine learning models to analyze the adsorption abilities and predict the capacity of loading the anticancer drug, 5-fluorouracil. Our findings indicate that different organic linkers, structural features, binding sites, topologies, <em>etc.</em> of COFs play an important role in determining the maximum loading capacity and release parameters of 5-FU. The implementation of molecular simulations-based machine learning methods for drug adsorption studies in COFs is rare in the literature. Once the model was validated, we studied the maximum loading capacity of 5-FU in a series of COFs, 102–108 and 112, from the COF database, as these exhibited a gradual trend in textural properties, aiming to understand this trend and the correlation between their structure and loading capacity. Then, we proceeded to study the adsorption process in detail in 4 of the COFs: three 2D COFs—COF-206, <em>i.e.</em>, D<small><sub>CuPc</sub></small>–A<small><sub>NDI</sub></small>-COF; COF-362, <em>i.e.</em>, PI-COF-3; and COF-398, <em>i.e.</em>, Py-DBA-COF-1—and one 3D COF—COF-363, <em>i.e.</em>, PI-COF-4. Radial distribution function and adsorption energy analyses revealed some important interactions and thermodynamic parameters leading to strong binding and slow release of 5-FU. The adsorption energy values in the top-performing COFs fall within the range of −8.43 to −42.25 × 10<small><sup>3</sup></small> kJ mol<small><sup>−1</sup></small>. The correlation of ML input parameters in terms of various chemical and structural descriptors with the maximum loading capacity is discussed. From the molecular simulations, COF-362 is the best-performing COF in terms of loading capacity and adsorption energy values. The ML models, <em>i.e.</em>, random forest, decision tree and three deep neural networks, were trained on 80% of the total data, while the remaining 20% of the data was used to test the models. DNN model-3 was chosen as the final model for further analysis based on <em>R</em><small><sup>2</sup></small> = 0.87, RMSE = 189.81, and MAE = 100.87. SHapley Additive exPlanations (SHAP) analysis and the feature importance chart indicated that among the structural descriptors, <em>S</em><small><sub>acc</sub></small>, LCD, and <em>V</em><small><sub>f</sub></small>, and among the chemical descriptors, C, H, and N, had the most positive impact on the output predictions of the model. Finally, a graphical user interface based on the best-performing ML model was created to predict the 5-FU loading capacity of COFs. This will save users time without the need to run the code or perform various tedious drug-loading experiments.</p>","PeriodicalId":101141,"journal":{"name":"RSC Pharmaceutics","volume":" 4","pages":" 703-717"},"PeriodicalIF":0.0000,"publicationDate":"2025-04-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.rsc.org/en/content/articlepdf/2025/pm/d5pm00007f?page=search","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"RSC Pharmaceutics","FirstCategoryId":"1085","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2025/pm/d5pm00007f","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
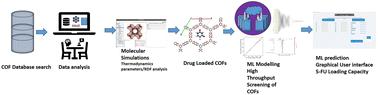
A database of 1242 experimentally synthesized COFs has been studied to understand their potential as drug carriers by employing molecular simulations and machine learning models to analyze the adsorption abilities and predict the capacity of loading the anticancer drug, 5-fluorouracil. Our findings indicate that different organic linkers, structural features, binding sites, topologies, etc. of COFs play an important role in determining the maximum loading capacity and release parameters of 5-FU. The implementation of molecular simulations-based machine learning methods for drug adsorption studies in COFs is rare in the literature. Once the model was validated, we studied the maximum loading capacity of 5-FU in a series of COFs, 102–108 and 112, from the COF database, as these exhibited a gradual trend in textural properties, aiming to understand this trend and the correlation between their structure and loading capacity. Then, we proceeded to study the adsorption process in detail in 4 of the COFs: three 2D COFs—COF-206, i.e., DCuPc–ANDI-COF; COF-362, i.e., PI-COF-3; and COF-398, i.e., Py-DBA-COF-1—and one 3D COF—COF-363, i.e., PI-COF-4. Radial distribution function and adsorption energy analyses revealed some important interactions and thermodynamic parameters leading to strong binding and slow release of 5-FU. The adsorption energy values in the top-performing COFs fall within the range of −8.43 to −42.25 × 103 kJ mol−1. The correlation of ML input parameters in terms of various chemical and structural descriptors with the maximum loading capacity is discussed. From the molecular simulations, COF-362 is the best-performing COF in terms of loading capacity and adsorption energy values. The ML models, i.e., random forest, decision tree and three deep neural networks, were trained on 80% of the total data, while the remaining 20% of the data was used to test the models. DNN model-3 was chosen as the final model for further analysis based on R2 = 0.87, RMSE = 189.81, and MAE = 100.87. SHapley Additive exPlanations (SHAP) analysis and the feature importance chart indicated that among the structural descriptors, Sacc, LCD, and Vf, and among the chemical descriptors, C, H, and N, had the most positive impact on the output predictions of the model. Finally, a graphical user interface based on the best-performing ML model was created to predict the 5-FU loading capacity of COFs. This will save users time without the need to run the code or perform various tedious drug-loading experiments.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


