Qichang Zhao, Haochen Zhao, Linyuan Guo, Kai Zheng, Yajie Li, Qiao Ling, Jing Tang, Yaohang Li, Jianxin Wang
{"title":"ColdstartCPI: Induced-fit theory-guided DTI predictive model with improved generalization performance","authors":"Qichang Zhao, Haochen Zhao, Linyuan Guo, Kai Zheng, Yajie Li, Qiao Ling, Jing Tang, Yaohang Li, Jianxin Wang","doi":"10.1038/s41467-025-61745-7","DOIUrl":null,"url":null,"abstract":"<p>Predicting compound-protein interactions (CPIs) plays a crucial role in drug discovery. Traditional methods, based on the key-lock theory and rigid docking, often fail with novel compounds and proteins due to their inability to account for molecular flexibility and the high sparsity of CPI data. Here, we introduce ColdstartCPI, a framework inspired by induced-fit theory, which leverages unsupervised pre-training features and a Transformer module to learn both compound and protein characteristics. ColdstartCPI treats proteins and compounds as flexible molecules during inference, aligning with biological insights. It outperforms state-of-the-art sequence-based models, particularly for unseen compounds and proteins, and shows strong generalization capability compared to structure-based methods in virtual screening. ColdstartCPI also excels in sparse and low-similarity data conditions, demonstrating its potential in data-limited settings. Our results are validated through literature search, molecular docking, and binding free energy calculations. Overall, ColdstartCPI offers a perspective on sequence-based drug design, presenting a promising tool for drug discovery.</p>","PeriodicalId":19066,"journal":{"name":"Nature Communications","volume":"11 1","pages":""},"PeriodicalIF":15.7000,"publicationDate":"2025-07-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Nature Communications","FirstCategoryId":"103","ListUrlMain":"https://doi.org/10.1038/s41467-025-61745-7","RegionNum":1,"RegionCategory":"综合性期刊","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MULTIDISCIPLINARY SCIENCES","Score":null,"Total":0}
引用次数: 0
Abstract
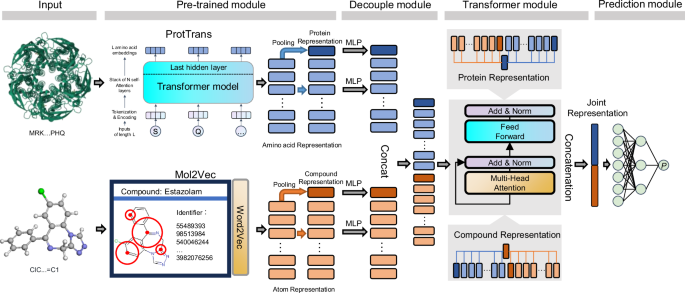
Predicting compound-protein interactions (CPIs) plays a crucial role in drug discovery. Traditional methods, based on the key-lock theory and rigid docking, often fail with novel compounds and proteins due to their inability to account for molecular flexibility and the high sparsity of CPI data. Here, we introduce ColdstartCPI, a framework inspired by induced-fit theory, which leverages unsupervised pre-training features and a Transformer module to learn both compound and protein characteristics. ColdstartCPI treats proteins and compounds as flexible molecules during inference, aligning with biological insights. It outperforms state-of-the-art sequence-based models, particularly for unseen compounds and proteins, and shows strong generalization capability compared to structure-based methods in virtual screening. ColdstartCPI also excels in sparse and low-similarity data conditions, demonstrating its potential in data-limited settings. Our results are validated through literature search, molecular docking, and binding free energy calculations. Overall, ColdstartCPI offers a perspective on sequence-based drug design, presenting a promising tool for drug discovery.
期刊介绍:
Nature Communications, an open-access journal, publishes high-quality research spanning all areas of the natural sciences. Papers featured in the journal showcase significant advances relevant to specialists in each respective field. With a 2-year impact factor of 16.6 (2022) and a median time of 8 days from submission to the first editorial decision, Nature Communications is committed to rapid dissemination of research findings. As a multidisciplinary journal, it welcomes contributions from biological, health, physical, chemical, Earth, social, mathematical, applied, and engineering sciences, aiming to highlight important breakthroughs within each domain.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


