{"title":"A novel intronic variant in the ASAH1 gene enhances aberrant splicing, causing spinal muscular atrophy with progressive myoclonic epilepsy.","authors":"Jinli Bai, Ping Li, Hui Jiao, Yuwei Jin, Hong Wang, Qinglin Jiang, Fang Song, Xiaoyin Peng, Yujin Qu","doi":"10.1186/s13052-025-02058-9","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Spinal muscular atrophy with progressive myoclonic epilepsy (SMA-PME) is a rare autosomal recessive disorder caused by ASAH1 gene variants. Although ASAH1 coding variants cause SMA-PME, the impact of noncoding variants, particularly noncanonical splice-site variants, is less clear.</p><p><strong>Methods: </strong>Whole-exome sequencing (WES) was performed on the proband, and Sanger sequencing was used to confirm the carrier status of the variants in the core family members. Complementary DNA (cDNA) and minigene splicing assays were performed to validate the splicing effects.</p><p><strong>Results: </strong>Two heterozygous ASAH1 variants were identified through WES: c.304dupA (p.Thr102Asnfs*14) and c.264 + 11A > G. Sanger sequencing confirmed that the variants were bi-parentally segregated in trans: c.304dupA was inherited from the father, and c.264 + 11A > G was inherited from the mother. The c.304dupA variant was classified as pathogenic according to the ACMG guidelines. However, the c.264 + 11A > G variant in intron 3 was reported for the first time, and its functional impact has not yet been fully elucidated. Complementary DNA (cDNA) and minigene splicing assays indicated that the c.264 + 11A > G variant generated two transcripts. Approximately 10% of the ASAH1 transcripts from the allele carrying c.264 + 11A > G were full length, whereas the remaining transcripts lacked exon 3. Exon skipping results from aberrant splicing, which potentially leads to a premature termination codon (PTC, p.Tyr59Ter).</p><p><strong>Conclusion: </strong>To the best of our knowledge, the c.264 + 11A > G is the first likely pathogenic noncanonical splice-site variant identified in this gene. This drives the pathogenesis of SMA-PME through exon 3 skipping. Our findings provide new insights into the intricate splicing mechanisms of noncanonical splice-site variants, emphasizing the unique role of cDNA analysis and minigene splicing assays in the precise diagnosis and genetic counseling of SMA-PME cases.</p>","PeriodicalId":14511,"journal":{"name":"Italian Journal of Pediatrics","volume":"51 1","pages":"214"},"PeriodicalIF":3.1000,"publicationDate":"2025-07-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12239438/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Italian Journal of Pediatrics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s13052-025-02058-9","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"PEDIATRICS","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Spinal muscular atrophy with progressive myoclonic epilepsy (SMA-PME) is a rare autosomal recessive disorder caused by ASAH1 gene variants. Although ASAH1 coding variants cause SMA-PME, the impact of noncoding variants, particularly noncanonical splice-site variants, is less clear.
Methods: Whole-exome sequencing (WES) was performed on the proband, and Sanger sequencing was used to confirm the carrier status of the variants in the core family members. Complementary DNA (cDNA) and minigene splicing assays were performed to validate the splicing effects.
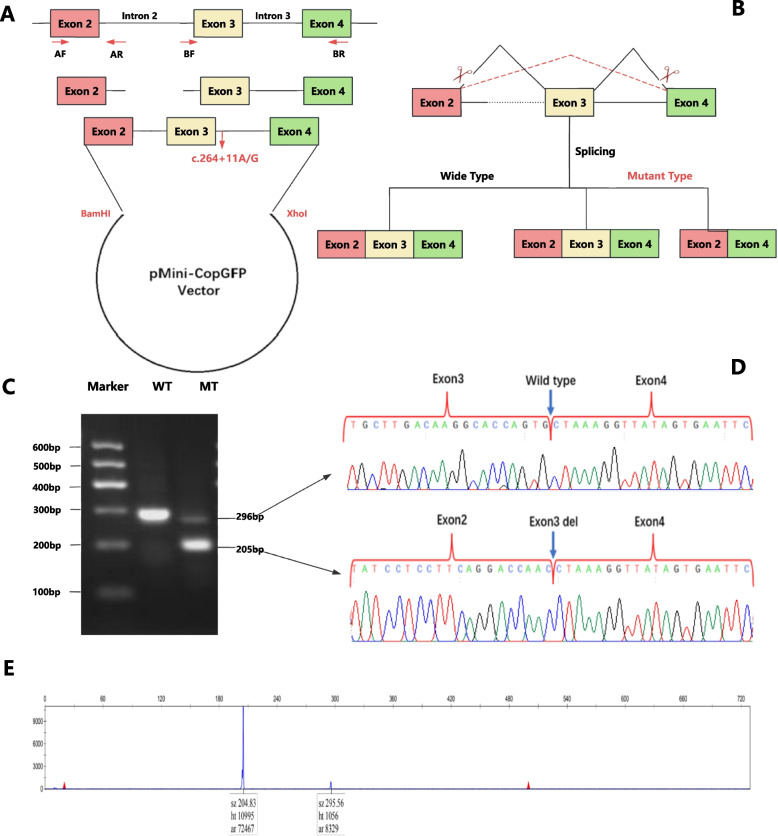
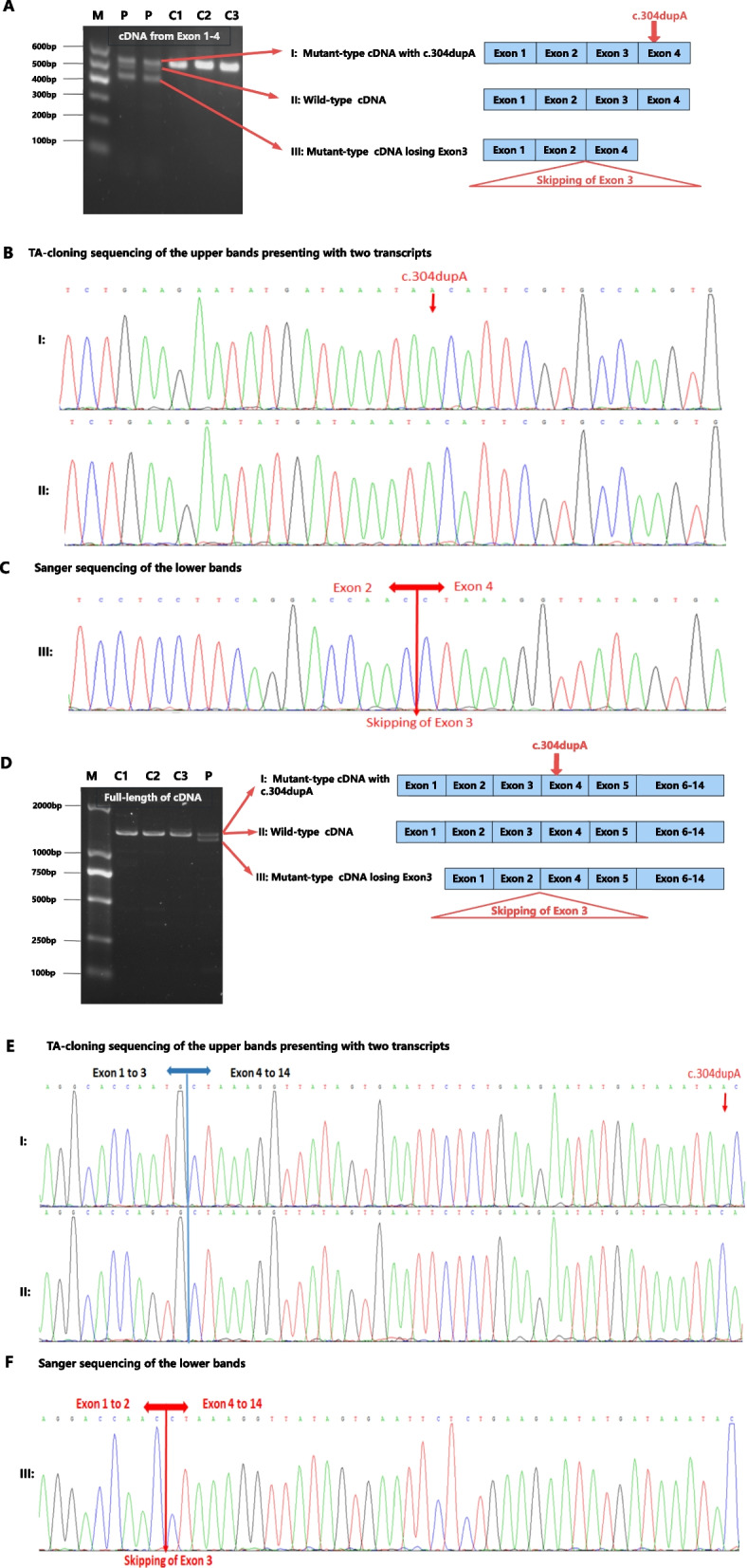
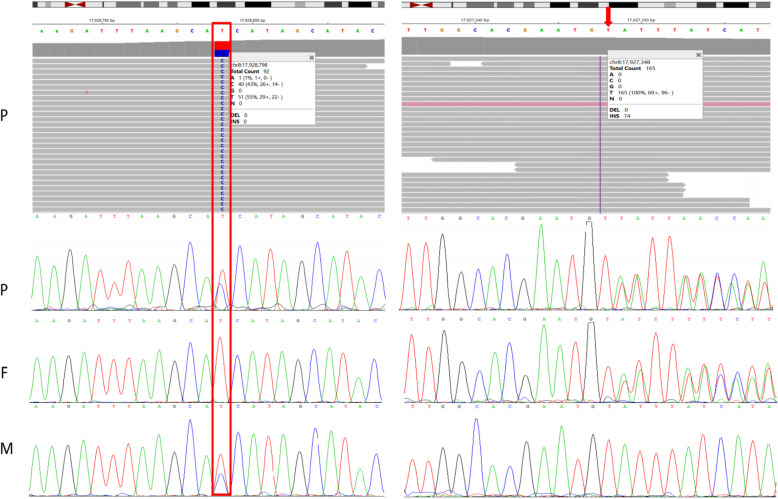
Results: Two heterozygous ASAH1 variants were identified through WES: c.304dupA (p.Thr102Asnfs*14) and c.264 + 11A > G. Sanger sequencing confirmed that the variants were bi-parentally segregated in trans: c.304dupA was inherited from the father, and c.264 + 11A > G was inherited from the mother. The c.304dupA variant was classified as pathogenic according to the ACMG guidelines. However, the c.264 + 11A > G variant in intron 3 was reported for the first time, and its functional impact has not yet been fully elucidated. Complementary DNA (cDNA) and minigene splicing assays indicated that the c.264 + 11A > G variant generated two transcripts. Approximately 10% of the ASAH1 transcripts from the allele carrying c.264 + 11A > G were full length, whereas the remaining transcripts lacked exon 3. Exon skipping results from aberrant splicing, which potentially leads to a premature termination codon (PTC, p.Tyr59Ter).
Conclusion: To the best of our knowledge, the c.264 + 11A > G is the first likely pathogenic noncanonical splice-site variant identified in this gene. This drives the pathogenesis of SMA-PME through exon 3 skipping. Our findings provide new insights into the intricate splicing mechanisms of noncanonical splice-site variants, emphasizing the unique role of cDNA analysis and minigene splicing assays in the precise diagnosis and genetic counseling of SMA-PME cases.
期刊介绍:
Italian Journal of Pediatrics is an open access peer-reviewed journal that includes all aspects of pediatric medicine. The journal also covers health service and public health research that addresses primary care issues.
The journal provides a high-quality forum for pediatricians and other healthcare professionals to report and discuss up-to-the-minute research and expert reviews in the field of pediatric medicine. The journal will continue to develop the range of articles published to enable this invaluable resource to stay at the forefront of the field.
Italian Journal of Pediatrics, which commenced in 1975 as Rivista Italiana di Pediatria, provides a high-quality forum for pediatricians and other healthcare professionals to report and discuss up-to-the-minute research and expert reviews in the field of pediatric medicine. The journal will continue to develop the range of articles published to enable this invaluable resource to stay at the forefront of the field.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


