{"title":"EGLN1-positive familial erythrocytosis: a rare variant with an unusually aggressive clinical course.","authors":"Laura Maule, Brielle Coe, Rishi Sawhney","doi":"10.1007/s12308-025-00645-7","DOIUrl":null,"url":null,"abstract":"<p><p>Familial erythrocytosis type 3 (ECYT3) is a rare condition caused by loss of function germline mutations in the prolyl hydroxylase domain-2 (PHD2), a regulator in the hypoxia-sensing pathway. Although mutations in PHD2 have been previously described, this particular variant lacks clinical characterization and presents with an aggressive course. We report the case of a patient with vasomotor symptoms and elevations in hematocrit (HCT) and hemoglobin (Hgb) despite frequent therapeutic phlebotomy. He had a family history of erythrocytosis spanning four generations. Germline genetic testing revealed a rare pathogenic variant of PHD2, confirming a diagnosis of ECYT3. Therapeutic phlebotomy yielded only transient Hgb and HCT reductions and only partial symptomatic control. This case highlights the diagnostic challenges and limitations of current treatments for hereditary erythrocytosis and underscores the need for symptom-centered management strategies. Furthermore, we highlight a gap in the literature around the pathophysiology and management of ECYT3.</p>","PeriodicalId":51320,"journal":{"name":"Journal of Hematopathology","volume":"18 1","pages":"30"},"PeriodicalIF":0.6000,"publicationDate":"2025-07-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12238123/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Hematopathology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s12308-025-00645-7","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"HEMATOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
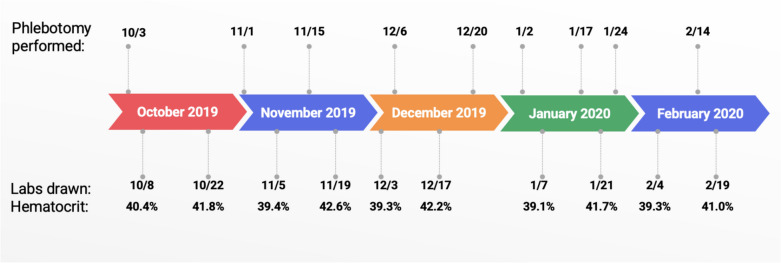
Familial erythrocytosis type 3 (ECYT3) is a rare condition caused by loss of function germline mutations in the prolyl hydroxylase domain-2 (PHD2), a regulator in the hypoxia-sensing pathway. Although mutations in PHD2 have been previously described, this particular variant lacks clinical characterization and presents with an aggressive course. We report the case of a patient with vasomotor symptoms and elevations in hematocrit (HCT) and hemoglobin (Hgb) despite frequent therapeutic phlebotomy. He had a family history of erythrocytosis spanning four generations. Germline genetic testing revealed a rare pathogenic variant of PHD2, confirming a diagnosis of ECYT3. Therapeutic phlebotomy yielded only transient Hgb and HCT reductions and only partial symptomatic control. This case highlights the diagnostic challenges and limitations of current treatments for hereditary erythrocytosis and underscores the need for symptom-centered management strategies. Furthermore, we highlight a gap in the literature around the pathophysiology and management of ECYT3.
期刊介绍:
The Journal of Hematopathology aims at providing pathologists with a special interest in hematopathology with all the information needed to perform modern pathology in evaluating lymphoid tissues and bone marrow. To this end the journal publishes reviews, editorials, comments, original papers, guidelines and protocols, papers on ancillary techniques, and occasional case reports in the fields of the pathology, molecular biology, and clinical features of diseases of the hematopoietic system.
The journal is the unique reference point for all pathologists with an interest in hematopathology. Molecular biologists involved in the expanding field of molecular diagnostics and research on lymphomas and leukemia benefit from the journal, too. Furthermore, the journal is of major interest for hematologists dealing with patients suffering from lymphomas, leukemias, and other diseases.
The journal is unique in its true international character. Especially in the field of hematopathology it is clear that there are huge geographical variations in incidence of diseases. This is not only locally relevant, but due to globalization, relevant for all those involved in the management of patients.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


