Zhenling Wang, Tianyi Shi, Weiliang Luo, Heather J. Kulik, Yang Liu, Xiaoye S. Li and Martin Head-Gordon*,
{"title":"Regularized Second-Order Møller–Plesset Theory: Linear Scaling Implementation and Assessment on Large-Molecule Problems","authors":"Zhenling Wang, Tianyi Shi, Weiliang Luo, Heather J. Kulik, Yang Liu, Xiaoye S. Li and Martin Head-Gordon*, ","doi":"10.1021/acs.jctc.5c00534","DOIUrl":null,"url":null,"abstract":"<p >Formally fifth-order scaling with molecular size, second-order Møller–Plesset (MP2) theory is widely used in its own right, and as a component of double hybrid density functional theory. Nevertheless, MP2 is well-known to have limited accuracy for large intermolecular interactions, and the MP2 energy is not bounded from below. Regularized MP2 methods aim to address these issues without changing the formal fifth-order scaling and are therefore of interest as more accurate and robust alternatives. In this work, we report a linear scaling algorithm with shared and distributed memory parallel implementations, for the evaluation of two regularized MP2 methods: κ-MP2 and the size-consistent second-order Brillouin–Wigner perturbation theory (BW-s2). The algorithm uses a single numerical threshold to control accuracy, which is demonstrated to yield a roughly 10-fold reduction in numerical error for each factor of 10 reduction in the threshold. The scaling of time-to-solution is assessed as a function of molecule size (up to 1800 atoms and 25000 atomic orbitals), the choice of threshold, and the number of processors (up to 2000 cores), for a range of systems including linear alkanes and three-dimensional complexes. The results show an earlier crossover against the canonical implementation for BW-s2 than for either MP2 or κ-MP2. The new implementation is used to evaluate the accuracy of the κ-MP2 and BW-s2 methods for some of the largest test sets of intermolecular interactions for which coupled cluster benchmark data are available, including IONPI19, L7, S12L, and C60ISO. The results show that κ-MP2 and BW-s2 perform far better than MP2. Additional tests of κ-MP2 and BW-s2 are reported for the case of short-range amino acid interactions, where MP2 performs exceptionally well.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":"21 14","pages":"6887–6904"},"PeriodicalIF":5.5000,"publicationDate":"2025-07-07","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jctc.5c00534","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
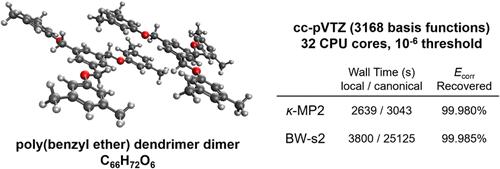
Formally fifth-order scaling with molecular size, second-order Møller–Plesset (MP2) theory is widely used in its own right, and as a component of double hybrid density functional theory. Nevertheless, MP2 is well-known to have limited accuracy for large intermolecular interactions, and the MP2 energy is not bounded from below. Regularized MP2 methods aim to address these issues without changing the formal fifth-order scaling and are therefore of interest as more accurate and robust alternatives. In this work, we report a linear scaling algorithm with shared and distributed memory parallel implementations, for the evaluation of two regularized MP2 methods: κ-MP2 and the size-consistent second-order Brillouin–Wigner perturbation theory (BW-s2). The algorithm uses a single numerical threshold to control accuracy, which is demonstrated to yield a roughly 10-fold reduction in numerical error for each factor of 10 reduction in the threshold. The scaling of time-to-solution is assessed as a function of molecule size (up to 1800 atoms and 25000 atomic orbitals), the choice of threshold, and the number of processors (up to 2000 cores), for a range of systems including linear alkanes and three-dimensional complexes. The results show an earlier crossover against the canonical implementation for BW-s2 than for either MP2 or κ-MP2. The new implementation is used to evaluate the accuracy of the κ-MP2 and BW-s2 methods for some of the largest test sets of intermolecular interactions for which coupled cluster benchmark data are available, including IONPI19, L7, S12L, and C60ISO. The results show that κ-MP2 and BW-s2 perform far better than MP2. Additional tests of κ-MP2 and BW-s2 are reported for the case of short-range amino acid interactions, where MP2 performs exceptionally well.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


