A novel USP27X missense variant identified in an individual with intellectual disability
IF 2.5
3区 生物学
Q2 GENETICS & HEREDITY
引用次数: 0
Abstract
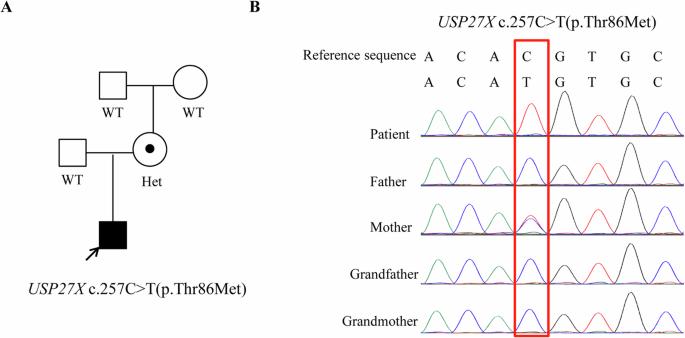
X-linked intellectual disability (XLID) is a group of neurodevelopmental disorders with genetic heterogeneity. Mutation of USP27X, a deubiquitinase encoding gene, is associated with X-linked intellectual developmental disorder-105 (XLID105), which is characterized by different combinations of impaired intellectual development (ID), developmental delay (DD), autism spectrum, attention deficit hyperactivity disorder and anxiety. Now only fourteen genetically diagnosed individuals have been reported. Here we describe a three-year boy with mild abnormal facial features, DD, severe speech delay and cognitive impairment, and ventricular septal defect. In addition, an increased nuchal translucency was observed during the fetal period. Trio whole-exome sequencing identified a novel missense variant, c.257 C > T (p.Thr86Met), in the USP27X gene (NM_001145073), which is inherited from his healthy mother and assessed to be a variant of uncertain significance. Further in vitro function study shows that this variant is detrimental to the expression and deubiquitination activity of USP27X. Our study provides more pathogenic evidences for this variant identified, and link this variant to the XLID-105 disease. In conclusion, our report expands the clinical and genetic spectrum of USP27X. Clinical trial registration: ChiCTR2000034358.
在智力残疾个体中发现了一种新的USP27X错义变体。
x连锁智力障碍(XLID)是一组具有遗传异质性的神经发育障碍。去泛素酶编码基因USP27X突变与x连锁智力发育障碍-105 (XLID105)有关,其特征是智力发育障碍(ID)、发育迟缓(DD)、自闭症谱系、注意缺陷多动障碍和焦虑的不同组合。现在只有14个基因诊断个体被报道。这里我们描述了一个三岁的男孩,轻度异常的面部特征,DD,严重的语言延迟和认知障碍,室间隔缺损。此外,在胎儿期观察到颈部透明度增加。三人全外显子组测序在USP27X基因(NM_001145073)中发现了一种新的错义变体C .257 C . > T (p.Thr86Met),该变异遗传自他的健康母亲,并被评估为一种不确定意义的变异。进一步的体外功能研究表明,该变异对USP27X的表达和去泛素化活性有害。我们的研究为该变异提供了更多的致病证据,并将该变异与XLID-105疾病联系起来。总之,我们的报告扩展了USP27X的临床和遗传谱。临床试验注册:ChiCTR2000034358。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Journal of Human Genetics
生物-遗传学
CiteScore
7.20
自引率
0.00%
发文量
101
审稿时长
4-8 weeks
期刊介绍:
The Journal of Human Genetics is an international journal publishing articles on human genetics, including medical genetics and human genome analysis. It covers all aspects of human genetics, including molecular genetics, clinical genetics, behavioral genetics, immunogenetics, pharmacogenomics, population genetics, functional genomics, epigenetics, genetic counseling and gene therapy.
Articles on the following areas are especially welcome: genetic factors of monogenic and complex disorders, genome-wide association studies, genetic epidemiology, cancer genetics, personal genomics, genotype-phenotype relationships and genome diversity.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


