Yaqin Zhang, Yu Xiong, Yuhang Wang, Qianqian Wang and Jun Fan
{"title":"Advances in computational approaches for bridging theory and experiments in electrocatalyst design","authors":"Yaqin Zhang, Yu Xiong, Yuhang Wang, Qianqian Wang and Jun Fan","doi":"10.1039/D5NH00216H","DOIUrl":null,"url":null,"abstract":"<p >The activation of inert molecules such as CO<small><sub>2</sub></small>, N<small><sub>2</sub></small>, and O<small><sub>2</sub></small> is central to addressing global energy and environmental challenges <em>via</em> electrocatalysis. However, their intrinsic stability and the complex solid–liquid interfacial phenomena present formidable obstacles for catalyst design. Recent advances in computational approaches are beginning to bridge the longstanding gap between idealized theoretical models and experimental realities. In this review, we highlight the progress made in scaling relations and descriptor-based screening methods, which underpin the Sabatier principle and volcano plot frameworks, enabling rapid identification of promising catalytic materials. We further discuss the evolution of thermodynamic and kinetic models—including the computational hydrogen electrode model, constant electrode potential model, and <em>ab initio</em> thermodynamics—that allow for accurate predictions of reaction energetics and catalyst stability under realistic operating conditions. Moreover, the advent of constant potential simulations and explicit solvation models, bolstered by <em>ab initio</em> molecular dynamics and machine learning-accelerated molecular dynamics, has significantly advanced our understanding of the dynamic electrochemical interface. High-throughput computational workflows and data-driven machine learning techniques have further streamlined catalyst discovery by efficiently exploring large material spaces and complex reaction pathways. Together, these computational advances not only provide mechanistic insights into inert molecule activation but also offer a robust platform for guiding experimental efforts. The review concludes with a discussion of remaining challenges and future opportunities to further integrate computational and experimental methodologies for the rational design of next-generation electrocatalysts.</p>","PeriodicalId":93,"journal":{"name":"Nanoscale Horizons","volume":" 10","pages":" 2211-2238"},"PeriodicalIF":6.6000,"publicationDate":"2025-07-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Nanoscale Horizons","FirstCategoryId":"88","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2025/nh/d5nh00216h","RegionNum":2,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
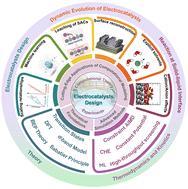
The activation of inert molecules such as CO2, N2, and O2 is central to addressing global energy and environmental challenges via electrocatalysis. However, their intrinsic stability and the complex solid–liquid interfacial phenomena present formidable obstacles for catalyst design. Recent advances in computational approaches are beginning to bridge the longstanding gap between idealized theoretical models and experimental realities. In this review, we highlight the progress made in scaling relations and descriptor-based screening methods, which underpin the Sabatier principle and volcano plot frameworks, enabling rapid identification of promising catalytic materials. We further discuss the evolution of thermodynamic and kinetic models—including the computational hydrogen electrode model, constant electrode potential model, and ab initio thermodynamics—that allow for accurate predictions of reaction energetics and catalyst stability under realistic operating conditions. Moreover, the advent of constant potential simulations and explicit solvation models, bolstered by ab initio molecular dynamics and machine learning-accelerated molecular dynamics, has significantly advanced our understanding of the dynamic electrochemical interface. High-throughput computational workflows and data-driven machine learning techniques have further streamlined catalyst discovery by efficiently exploring large material spaces and complex reaction pathways. Together, these computational advances not only provide mechanistic insights into inert molecule activation but also offer a robust platform for guiding experimental efforts. The review concludes with a discussion of remaining challenges and future opportunities to further integrate computational and experimental methodologies for the rational design of next-generation electrocatalysts.
期刊介绍:
Nanoscale Horizons stands out as a premier journal for publishing exceptionally high-quality and innovative nanoscience and nanotechnology. The emphasis lies on original research that introduces a new concept or a novel perspective (a conceptual advance), prioritizing this over reporting technological improvements. Nevertheless, outstanding articles showcasing truly groundbreaking developments, including record-breaking performance, may also find a place in the journal. Published work must be of substantial general interest to our broad and diverse readership across the nanoscience and nanotechnology community.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


