Molecular Modeling Study for the Design of New FGFR4 Inhibitors Using 3D‑QSAR, Molecular Docking, Molecular Dynamic, ADMET Prediction, and Retrosynthesis
Jun Li, Guangyi Luo, Qiuyu Xiong, Xinyi Chen, Zhengyang Zhao, Pengcheng Wen, Dr. Lu Zheng, Dr. Qingkun Wu
{"title":"Molecular Modeling Study for the Design of New FGFR4 Inhibitors Using 3D‑QSAR, Molecular Docking, Molecular Dynamic, ADMET Prediction, and Retrosynthesis","authors":"Jun Li, Guangyi Luo, Qiuyu Xiong, Xinyi Chen, Zhengyang Zhao, Pengcheng Wen, Dr. Lu Zheng, Dr. Qingkun Wu","doi":"10.1002/slct.202502220","DOIUrl":null,"url":null,"abstract":"<p>Fibroblast growth factor receptor 4 (FGFR4), a receptor tyrosine kinase, has emerged as a promising therapeutic target for liver cancer. In this study, we present a novel structure-based drug design approach combining computational techniques to develop quinazoline-based FGFR4 inhibitors. Our innovative strategy employed comparative molecular field analysis (CoMFA) and comparative molecular similarity index analysis (CoMSIA) to establish highly predictive 3D-QSAR models (CoMFA: <i>q</i><sup>2</sup> = 0.552, <i>r</i><sup>2</sup> = 0.997; CoMSIA: <i>q</i><sup>2</sup> = 0.618, <i>r</i><sup>2</sup> = 0.960), revealing critical structure-activity relationships. Building on these insights, we designed a series of novel quinazoline derivatives featuring optimized pharmacophoric elements. Notably, our lead compound <b>N8</b> demonstrated: (1) superior binding stability in molecular docking studies, (2) enhanced predicted inhibitory activity, and (3) improved ADMET properties compared to existing inhibitors. Molecular dynamics simulations (100 ns) confirmed the remarkable stability of the N8-FGFR4 complex, validating our design strategy. Furthermore, a practical retrosynthetic pathway was developed for new compounds, addressing previous synthetic challenges and facilitating future development. This work not only develops a promising drug candidate but also establishes a robust framework for designing FGFR4-targeted therapies, ensuring a seamless progression of subsequent research endeavors.</p>","PeriodicalId":146,"journal":{"name":"ChemistrySelect","volume":"10 25","pages":""},"PeriodicalIF":2.0000,"publicationDate":"2025-07-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"ChemistrySelect","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/slct.202502220","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
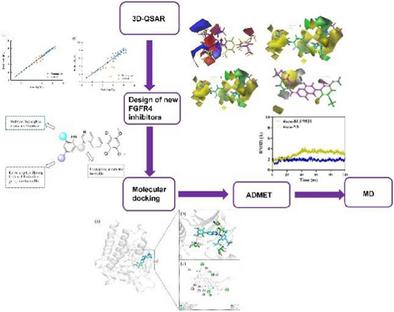
Fibroblast growth factor receptor 4 (FGFR4), a receptor tyrosine kinase, has emerged as a promising therapeutic target for liver cancer. In this study, we present a novel structure-based drug design approach combining computational techniques to develop quinazoline-based FGFR4 inhibitors. Our innovative strategy employed comparative molecular field analysis (CoMFA) and comparative molecular similarity index analysis (CoMSIA) to establish highly predictive 3D-QSAR models (CoMFA: q2 = 0.552, r2 = 0.997; CoMSIA: q2 = 0.618, r2 = 0.960), revealing critical structure-activity relationships. Building on these insights, we designed a series of novel quinazoline derivatives featuring optimized pharmacophoric elements. Notably, our lead compound N8 demonstrated: (1) superior binding stability in molecular docking studies, (2) enhanced predicted inhibitory activity, and (3) improved ADMET properties compared to existing inhibitors. Molecular dynamics simulations (100 ns) confirmed the remarkable stability of the N8-FGFR4 complex, validating our design strategy. Furthermore, a practical retrosynthetic pathway was developed for new compounds, addressing previous synthetic challenges and facilitating future development. This work not only develops a promising drug candidate but also establishes a robust framework for designing FGFR4-targeted therapies, ensuring a seamless progression of subsequent research endeavors.
期刊介绍:
ChemistrySelect is the latest journal from ChemPubSoc Europe and Wiley-VCH. It offers researchers a quality society-owned journal in which to publish their work in all areas of chemistry. Manuscripts are evaluated by active researchers to ensure they add meaningfully to the scientific literature, and those accepted are processed quickly to ensure rapid online publication.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


