Xin Chen, Li Tai Fang, Zhong Chen, Wanqiu Chen, Hongjin Wu, Bin Zhu, Malcolm Moos, Andrew Farmer, Xiaowen Zhang, Wei Xiong, Shusheng Gong, Wendell Jones, Christopher E Mason, Shixiu Wu, Chunlin Xiao, Charles Wang
{"title":"A benchmarking study of copy number variation inference methods using single-cell RNA-sequencing data.","authors":"Xin Chen, Li Tai Fang, Zhong Chen, Wanqiu Chen, Hongjin Wu, Bin Zhu, Malcolm Moos, Andrew Farmer, Xiaowen Zhang, Wei Xiong, Shusheng Gong, Wendell Jones, Christopher E Mason, Shixiu Wu, Chunlin Xiao, Charles Wang","doi":"10.1093/pcmedi/pbaf011","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Single-cell RNA-sequencing (scRNA-seq) has emerged as a powerful tool for cancer research, enabling in-depth characterization of tumor heterogeneity at the single-cell level. Recently, several scRNA-seq copy number variation (scCNV) inference methods have been developed, expanding the application of scRNA-seq to study genetic heterogeneity in cancer using transcriptomic data. However, the fidelity of these methods has not been investigated systematically.</p><p><strong>Methods: </strong>We benchmarked five commonly used scCNV inference methods: HoneyBADGER, CopyKAT, CaSpER, inferCNV, and sciCNV. We evaluated their performance across four different scRNA-seq platforms using data from our previous multicenter study. We evaluated scCNV performance further using scRNA-seq datasets derived from mixed samples consisting of five human lung adenocarcinoma cell lines and also sequenced tissues from a small cell lung cancer patient and used the data to validate our findings with a clinical scRNA-seq dataset.</p><p><strong>Results: </strong>We found that the sensitivity and specificity of the five scCNV inference methods varied, depending on the selection of reference data, sequencing depth, and read length. CopyKAT and CaSpER outperformed other methods overall, while inferCNV, sciCNV, and CopyKAT performed better than other methods in subclone identification. We found that batch effects significantly affected the performance of subclone identification in mixed datasets in most methods we tested.</p><p><strong>Conclusion: </strong>Our benchmarking study revealed the strengths and weaknesses of each of these scCNV inference methods and provided guidance for selecting the optimal CNV inference method using scRNA-seq data.</p>","PeriodicalId":33608,"journal":{"name":"Precision Clinical Medicine","volume":"8 2","pages":"pbaf011"},"PeriodicalIF":5.0000,"publicationDate":"2025-06-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12204187/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Precision Clinical Medicine","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1093/pcmedi/pbaf011","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/6/1 0:00:00","PubModel":"eCollection","JCR":"Q1","JCRName":"MEDICINE, RESEARCH & EXPERIMENTAL","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Single-cell RNA-sequencing (scRNA-seq) has emerged as a powerful tool for cancer research, enabling in-depth characterization of tumor heterogeneity at the single-cell level. Recently, several scRNA-seq copy number variation (scCNV) inference methods have been developed, expanding the application of scRNA-seq to study genetic heterogeneity in cancer using transcriptomic data. However, the fidelity of these methods has not been investigated systematically.
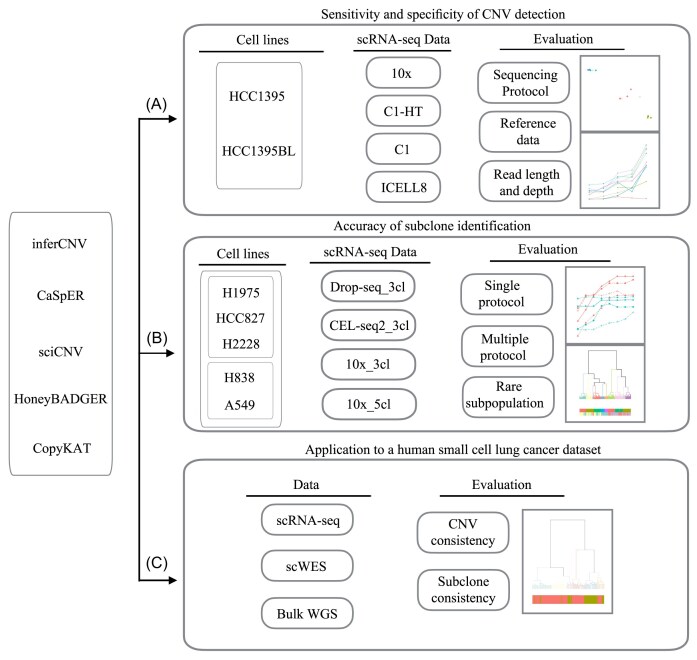
Methods: We benchmarked five commonly used scCNV inference methods: HoneyBADGER, CopyKAT, CaSpER, inferCNV, and sciCNV. We evaluated their performance across four different scRNA-seq platforms using data from our previous multicenter study. We evaluated scCNV performance further using scRNA-seq datasets derived from mixed samples consisting of five human lung adenocarcinoma cell lines and also sequenced tissues from a small cell lung cancer patient and used the data to validate our findings with a clinical scRNA-seq dataset.
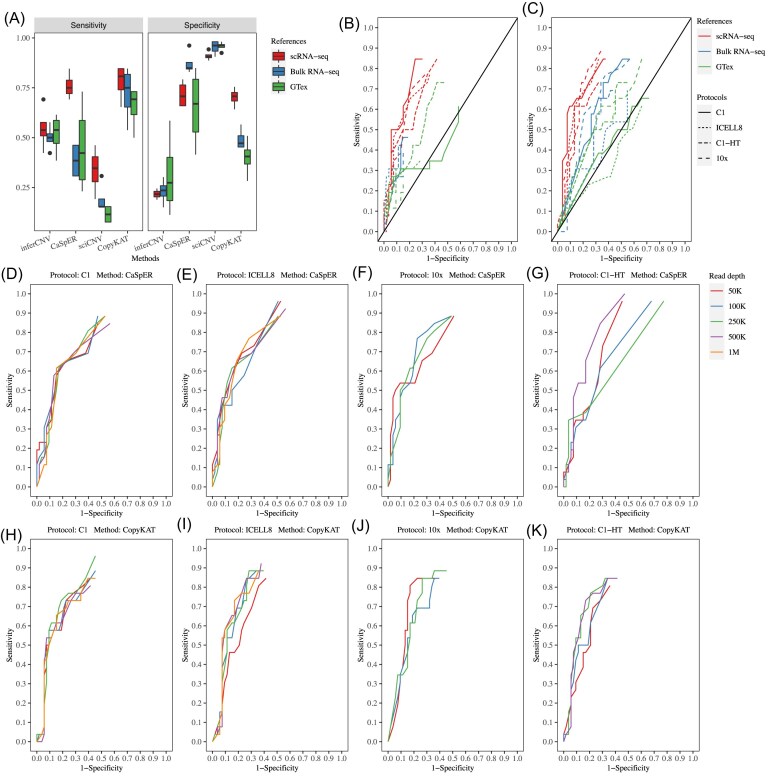
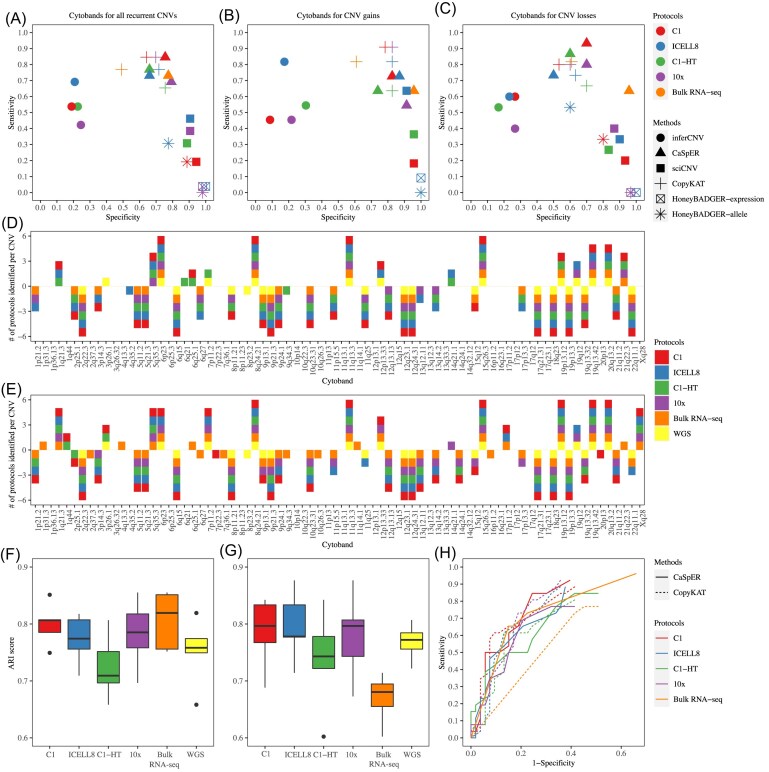
Results: We found that the sensitivity and specificity of the five scCNV inference methods varied, depending on the selection of reference data, sequencing depth, and read length. CopyKAT and CaSpER outperformed other methods overall, while inferCNV, sciCNV, and CopyKAT performed better than other methods in subclone identification. We found that batch effects significantly affected the performance of subclone identification in mixed datasets in most methods we tested.
Conclusion: Our benchmarking study revealed the strengths and weaknesses of each of these scCNV inference methods and provided guidance for selecting the optimal CNV inference method using scRNA-seq data.
期刊介绍:
Precision Clinical Medicine (PCM) is an international, peer-reviewed, open access journal that provides timely publication of original research articles, case reports, reviews, editorials, and perspectives across the spectrum of precision medicine. The journal's mission is to deliver new theories, methods, and evidence that enhance disease diagnosis, treatment, prevention, and prognosis, thereby establishing a vital communication platform for clinicians and researchers that has the potential to transform medical practice. PCM encompasses all facets of precision medicine, which involves personalized approaches to diagnosis, treatment, and prevention, tailored to individual patients or patient subgroups based on their unique genetic, phenotypic, or psychosocial profiles. The clinical conditions addressed by the journal include a wide range of areas such as cancer, infectious diseases, inherited diseases, complex diseases, and rare diseases.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


