Peter Reinholdt*, Karl Michael Ziems, Erik Rosendahl Kjellgren, Sonia Coriani, Stephan P. A. Sauer and Jacob Kongsted,
{"title":"Critical Limitations in Quantum-Selected Configuration Interaction Methods","authors":"Peter Reinholdt*, Karl Michael Ziems, Erik Rosendahl Kjellgren, Sonia Coriani, Stephan P. A. Sauer and Jacob Kongsted, ","doi":"10.1021/acs.jctc.5c00375","DOIUrl":null,"url":null,"abstract":"<p >Quantum Selected Configuration Interaction (QSCI) methods (also known as Sample-based Quantum Diagonalization, SQD) have emerged as promising near-term approaches to solving the electronic Schrödinger equation with quantum computers. In this work, we perform numerical analysis to show that QSCI methods face critical limitations that severely hinder their practical applicability in chemistry. Using the nitrogen molecule and the iron–sulfur cluster [2Fe–2S] as examples, we demonstrate that while QSCI can, in principle, yield high-quality configuration interaction (CI) expansions similar to classical SCI heuristics in some cases, the method struggles with inefficiencies in finding new determinants as sampling repeatedly selects already seen configurations. This inefficiency becomes especially pronounced when targeting high-accuracy results or sampling from an approximate ansatz. In cases where the sampling problem is not present, the resulting CI expansions are less compact than those generated from classical heuristics, rendering QSCI an overall more expensive method. Our findings suggest a significant drawback in QSCI methods when sampling from the ground-state distribution as the inescapable trade-off between finding sufficiently many determinants and generating compact, accurate CI expansions. This ultimately hinders utility in quantum chemistry applications, as QSCI falls behind more efficient classical counterparts.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":"21 14","pages":"6811–6822"},"PeriodicalIF":5.5000,"publicationDate":"2025-06-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jctc.5c00375","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
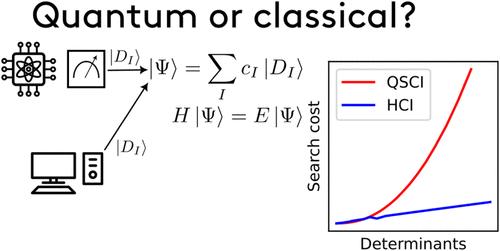
Quantum Selected Configuration Interaction (QSCI) methods (also known as Sample-based Quantum Diagonalization, SQD) have emerged as promising near-term approaches to solving the electronic Schrödinger equation with quantum computers. In this work, we perform numerical analysis to show that QSCI methods face critical limitations that severely hinder their practical applicability in chemistry. Using the nitrogen molecule and the iron–sulfur cluster [2Fe–2S] as examples, we demonstrate that while QSCI can, in principle, yield high-quality configuration interaction (CI) expansions similar to classical SCI heuristics in some cases, the method struggles with inefficiencies in finding new determinants as sampling repeatedly selects already seen configurations. This inefficiency becomes especially pronounced when targeting high-accuracy results or sampling from an approximate ansatz. In cases where the sampling problem is not present, the resulting CI expansions are less compact than those generated from classical heuristics, rendering QSCI an overall more expensive method. Our findings suggest a significant drawback in QSCI methods when sampling from the ground-state distribution as the inescapable trade-off between finding sufficiently many determinants and generating compact, accurate CI expansions. This ultimately hinders utility in quantum chemistry applications, as QSCI falls behind more efficient classical counterparts.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


