Machine learning prediction of intestinal α-glucosidase inhibitors using a diverse set of ligands: a drug repurposing effort with drugBank database screening.
Adeshina I Odugbemi, Clement Nyirenda, Alan Christoffels, Samuel A Egieyeh
{"title":"Machine learning prediction of intestinal α-glucosidase inhibitors using a diverse set of ligands: a drug repurposing effort with drugBank database screening.","authors":"Adeshina I Odugbemi, Clement Nyirenda, Alan Christoffels, Samuel A Egieyeh","doi":"10.1007/s40203-025-00384-8","DOIUrl":null,"url":null,"abstract":"<p><p>The global rise in diabetes mellitus (DM) poses a significant health challenge, necessitating effective therapeutic interventions. α-Glucosidase inhibitors play a crucial role in managing postprandial hyperglycemia and reducing the risk of complications in Type 2 DM. Quantitative Structure-Activity Relationship (QSAR) modelling is critical in computational drug discovery. However, many QSAR studies on α-glucosidase inhibitors often rely on limited compound series and statistical methods, restricting their applicability across wide chemical space. Integrating machine learning (ML) into QSAR offers a promising avenue for discovering novel therapeutic compounds by handling complex information from diverse compound sets. Our study aimed to develop robust predictive models for α-glucosidase inhibitors using a dataset of 1082 compounds with known activity against intestinal α-glucosidase (maltase-glucoamylase). After data preparation, we used 626 compounds to train ML models, generating different training data of three distinct molecular representations: 2D-descriptors, 3D-descriptors, and Extended-connectivity-fingerprint (ECFP4). These models, trained on random forest and support vector machine algorithms, underwent rigorous evaluation using established metrics. Subsequently, the best-performing model was used to screen the Drugbank database, identifying potential α-glucosidase inhibitor drugs. Drug repurposing, an expedited strategy for identifying new therapeutic uses for existing drugs, holds immense potential in this regard. Molecular docking and molecular dynamics simulations further corroborated our predictions. Our results indicate that 2D descriptors and ECFP4 molecular representations outperform 3D descriptors. Furthermore, drug candidates identified from DrugBank screening exhibited promising binding interactions with α-glucosidase, supporting our ML predictions and their potential for drug repurposing.</p><p><strong>Supplementary information: </strong>The online version contains supplementary material available at 10.1007/s40203-025-00384-8.</p>","PeriodicalId":94038,"journal":{"name":"In silico pharmacology","volume":"13 2","pages":"95"},"PeriodicalIF":0.0000,"publicationDate":"2025-06-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12198089/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"In silico pharmacology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1007/s40203-025-00384-8","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
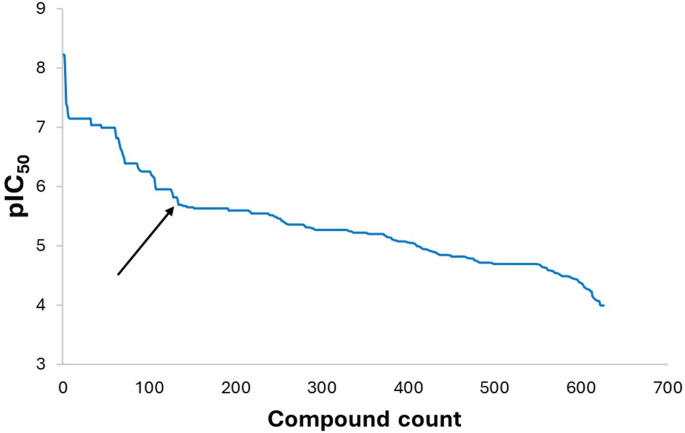
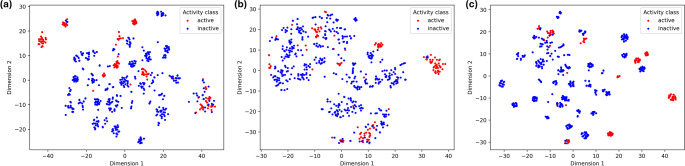
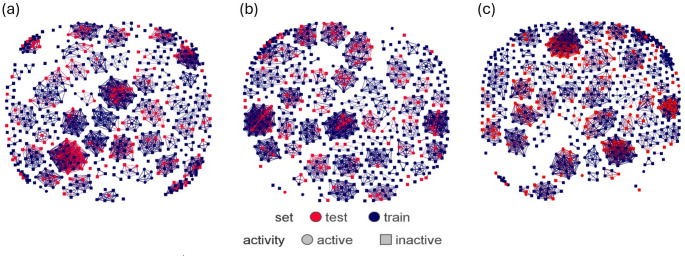
The global rise in diabetes mellitus (DM) poses a significant health challenge, necessitating effective therapeutic interventions. α-Glucosidase inhibitors play a crucial role in managing postprandial hyperglycemia and reducing the risk of complications in Type 2 DM. Quantitative Structure-Activity Relationship (QSAR) modelling is critical in computational drug discovery. However, many QSAR studies on α-glucosidase inhibitors often rely on limited compound series and statistical methods, restricting their applicability across wide chemical space. Integrating machine learning (ML) into QSAR offers a promising avenue for discovering novel therapeutic compounds by handling complex information from diverse compound sets. Our study aimed to develop robust predictive models for α-glucosidase inhibitors using a dataset of 1082 compounds with known activity against intestinal α-glucosidase (maltase-glucoamylase). After data preparation, we used 626 compounds to train ML models, generating different training data of three distinct molecular representations: 2D-descriptors, 3D-descriptors, and Extended-connectivity-fingerprint (ECFP4). These models, trained on random forest and support vector machine algorithms, underwent rigorous evaluation using established metrics. Subsequently, the best-performing model was used to screen the Drugbank database, identifying potential α-glucosidase inhibitor drugs. Drug repurposing, an expedited strategy for identifying new therapeutic uses for existing drugs, holds immense potential in this regard. Molecular docking and molecular dynamics simulations further corroborated our predictions. Our results indicate that 2D descriptors and ECFP4 molecular representations outperform 3D descriptors. Furthermore, drug candidates identified from DrugBank screening exhibited promising binding interactions with α-glucosidase, supporting our ML predictions and their potential for drug repurposing.
Supplementary information: The online version contains supplementary material available at 10.1007/s40203-025-00384-8.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


