Khalid Omeir, Jacob Ancira, Rebecca Gabrilska, Craig Tipton, Clint Miller, Ashley Noe, Kumudu Subasinghe, Megan Rowe, Nicole Phillips, Joseph Wolcott, Caleb D Philips
{"title":"Heritable Tissue-Specific Gene Expression Associates With Chronic Wound Microbial Species.","authors":"Khalid Omeir, Jacob Ancira, Rebecca Gabrilska, Craig Tipton, Clint Miller, Ashley Noe, Kumudu Subasinghe, Megan Rowe, Nicole Phillips, Joseph Wolcott, Caleb D Philips","doi":"10.1111/wrr.70055","DOIUrl":null,"url":null,"abstract":"<p><p>The reasons for interpatient variability in chronic wound microbiome composition are thought to be complex but are poorly known. To investigate how patients' genetically regulated tissue expression may influence chronic wound bacterial composition, we performed a microbiome-transcriptome-wide association study. This approach involved estimating for 509 patients their tissue-specific gene expression from DNA genotypes, followed by associating gene expression to the relative abundances of species detected in their wounds as provided on clinical reports to the physician. Comparisons to artery, blood, fibroblast, skeletal muscle, skin, subcutaneous fat, and nerve tissue resulted in 251 transcriptional differences at 109 genes significantly explaining abundances of 39 different species. Overall, these species were detected in ~63% of wounds. A similar number of associations per tissue was observed (range 31-39), and many genes were associated at multiple tissues in distinct ways. The cumulative variance across loci for species relative abundance explained ranged from ~5%-36%, depending on species. Although the same gene was almost never associated with more than one species, ~14% of enriched pathways were independently enriched for multiple species, which may reflect the diversity of ways microbes interact with partially overlapping attributes of the wound bed. Commonly enriched pathways pertained to collagen formation and modification, cell signalling, cytoskeletal dynamics, interactions with extracellular matrix, transmembrane proteins, amongst others. This work expands the new perspective that individual genetics may partially determine microbial colonisation and infection.</p>","PeriodicalId":23864,"journal":{"name":"Wound Repair and Regeneration","volume":"33 4","pages":"e70055"},"PeriodicalIF":3.4000,"publicationDate":"2025-07-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12203766/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Wound Repair and Regeneration","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1111/wrr.70055","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CELL BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
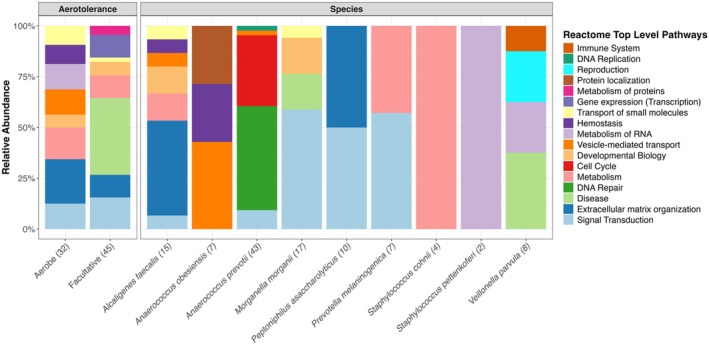
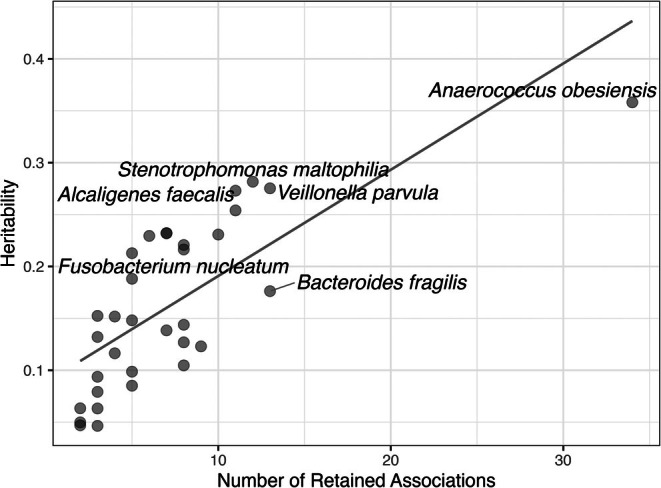
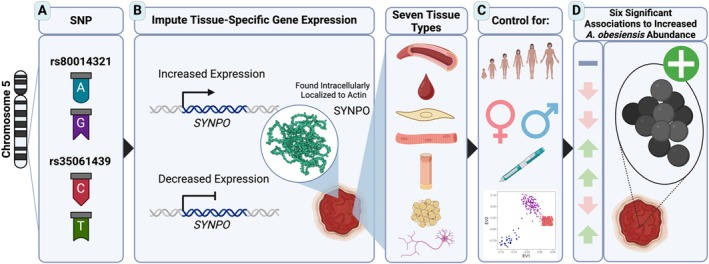
The reasons for interpatient variability in chronic wound microbiome composition are thought to be complex but are poorly known. To investigate how patients' genetically regulated tissue expression may influence chronic wound bacterial composition, we performed a microbiome-transcriptome-wide association study. This approach involved estimating for 509 patients their tissue-specific gene expression from DNA genotypes, followed by associating gene expression to the relative abundances of species detected in their wounds as provided on clinical reports to the physician. Comparisons to artery, blood, fibroblast, skeletal muscle, skin, subcutaneous fat, and nerve tissue resulted in 251 transcriptional differences at 109 genes significantly explaining abundances of 39 different species. Overall, these species were detected in ~63% of wounds. A similar number of associations per tissue was observed (range 31-39), and many genes were associated at multiple tissues in distinct ways. The cumulative variance across loci for species relative abundance explained ranged from ~5%-36%, depending on species. Although the same gene was almost never associated with more than one species, ~14% of enriched pathways were independently enriched for multiple species, which may reflect the diversity of ways microbes interact with partially overlapping attributes of the wound bed. Commonly enriched pathways pertained to collagen formation and modification, cell signalling, cytoskeletal dynamics, interactions with extracellular matrix, transmembrane proteins, amongst others. This work expands the new perspective that individual genetics may partially determine microbial colonisation and infection.
期刊介绍:
Wound Repair and Regeneration provides extensive international coverage of cellular and molecular biology, connective tissue, and biological mediator studies in the field of tissue repair and regeneration and serves a diverse audience of surgeons, plastic surgeons, dermatologists, biochemists, cell biologists, and others.
Wound Repair and Regeneration is the official journal of The Wound Healing Society, The European Tissue Repair Society, The Japanese Society for Wound Healing, and The Australian Wound Management Association.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


