Yuehua Dong, Shaohua Pei, Zirao Yang, Yanyan Xue, He Wang
{"title":"A novel variant in NPR2: C.2291T > C (p.Leu764Pro) identified in a patient with acromesomelic dysplasia Maroteaux type.","authors":"Yuehua Dong, Shaohua Pei, Zirao Yang, Yanyan Xue, He Wang","doi":"10.1186/s13052-025-02050-3","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Acromesomelic dysplasia Maroteaux type (AMDM) is a rare autosomal recessive skeletal dysplasia with an estimated prevalence of 1:1,000,000. It is characterized by extreme shortening of the forelimbs and disproportionate short stature.</p><p><strong>Case presentation: </strong>Here we present the clinical and genetic features of an 8-year-8-month-old boy exhibiting idiopathic short stature and abnormal changes of the appendicular skeleton and axial skeleton, consistent with the established phenotypic spectrum of AMDM. Using diagnostic exome sequencing, we identified two variants in NPR2: a known pathogenic nonsense variant, C.2965 C > T (p.Arg989*), and a missense variant of unknown significance, C.2291T > C (p.Leu764Pro), which has never been reported before. Sanger sequencing confirmed that the variants were inherited from his phenotypically normal parents. The proband is compound heterozygous, while both parents are heterozygous carriers, indicating an autosomal recessive pattern of inheritance.</p><p><strong>Conclusion: </strong>This study enriches the pathogenic gene mutation spectrum of NPR2 in patients with AMDM and further emphasizes the application of molecular genetic detection in the diagnosis of rare skeletal abnormalities.</p>","PeriodicalId":14511,"journal":{"name":"Italian Journal of Pediatrics","volume":"51 1","pages":"199"},"PeriodicalIF":3.1000,"publicationDate":"2025-06-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12186393/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Italian Journal of Pediatrics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s13052-025-02050-3","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"PEDIATRICS","Score":null,"Total":0}
引用次数: 0
Abstract
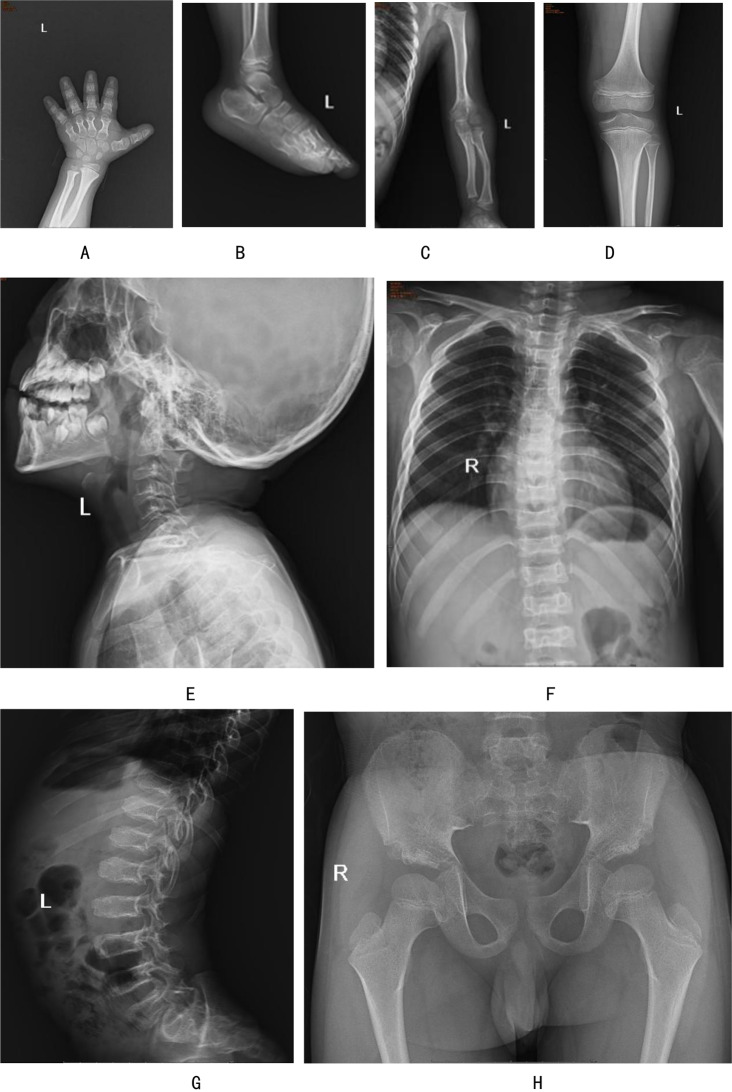
Background: Acromesomelic dysplasia Maroteaux type (AMDM) is a rare autosomal recessive skeletal dysplasia with an estimated prevalence of 1:1,000,000. It is characterized by extreme shortening of the forelimbs and disproportionate short stature.
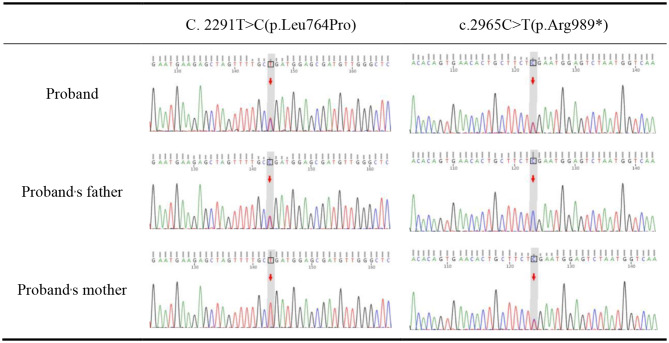
Case presentation: Here we present the clinical and genetic features of an 8-year-8-month-old boy exhibiting idiopathic short stature and abnormal changes of the appendicular skeleton and axial skeleton, consistent with the established phenotypic spectrum of AMDM. Using diagnostic exome sequencing, we identified two variants in NPR2: a known pathogenic nonsense variant, C.2965 C > T (p.Arg989*), and a missense variant of unknown significance, C.2291T > C (p.Leu764Pro), which has never been reported before. Sanger sequencing confirmed that the variants were inherited from his phenotypically normal parents. The proband is compound heterozygous, while both parents are heterozygous carriers, indicating an autosomal recessive pattern of inheritance.
Conclusion: This study enriches the pathogenic gene mutation spectrum of NPR2 in patients with AMDM and further emphasizes the application of molecular genetic detection in the diagnosis of rare skeletal abnormalities.
在Maroteaux型端端粒发育不良患者中发现了一种新的NPR2变异:C. 2291t > C (p.Leu764Pro)。
背景:马罗多型肢端粒发育不良(AMDM)是一种罕见的常染色体隐性骨骼发育不良,估计患病率为1:100万。它的特点是前肢极度缩短,身材矮小不成比例。病例报告:在这里,我们报告了一个8岁-8个月大的男孩的临床和遗传特征,表现为特发性身材矮小和阑尾骨骼和轴向骨骼的异常变化,与已建立的AMDM表型谱一致。通过诊断外显子组测序,我们鉴定出了NPR2的两个变异:一个已知的致病无义变异,C.2965 C > T (p.a g989*),以及一个未知意义的错义变异,C. 2291t > C (p.l u764pro),这在以前从未报道过。桑格测序证实,这些变异遗传自他表型正常的父母。先证者为复合杂合型,父母双方均为杂合型携带者,为常染色体隐性遗传模式。结论:本研究丰富了AMDM患者NPR2致病基因突变谱,进一步强调了分子遗传学检测在罕见骨骼异常诊断中的应用。
期刊介绍:
Italian Journal of Pediatrics is an open access peer-reviewed journal that includes all aspects of pediatric medicine. The journal also covers health service and public health research that addresses primary care issues.
The journal provides a high-quality forum for pediatricians and other healthcare professionals to report and discuss up-to-the-minute research and expert reviews in the field of pediatric medicine. The journal will continue to develop the range of articles published to enable this invaluable resource to stay at the forefront of the field.
Italian Journal of Pediatrics, which commenced in 1975 as Rivista Italiana di Pediatria, provides a high-quality forum for pediatricians and other healthcare professionals to report and discuss up-to-the-minute research and expert reviews in the field of pediatric medicine. The journal will continue to develop the range of articles published to enable this invaluable resource to stay at the forefront of the field.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


