Maura Mancini, Paola Palino, Alessandro Calderone, Giovanni W Oliverio, Pasquale Aragona, Alessandro Meduri
{"title":"Coexistence of Congenital Aniridia and Ptosis in a Patient with Neurofibromatosis Type I: A Case Report.","authors":"Maura Mancini, Paola Palino, Alessandro Calderone, Giovanni W Oliverio, Pasquale Aragona, Alessandro Meduri","doi":"10.1159/000546420","DOIUrl":null,"url":null,"abstract":"<p><strong>Introduction: </strong>Neurofibromatosis type 1 (NF1) is a genetic disorder caused by mutations in the <i>NF1</i> gene on chromosome 17q11.2. The main ocular manifestations include Lisch nodules, optic pathway gliomas, and plexiform neurofibromas, all of which can potentially impair visual function. Despite the numerous documented ocular manifestations of NF1, congenital aniridia has never been previously reported. Aniridia is a rare congenital disorder primarily associated with mutations in the <i>PAX6</i> gene, leading to iris hypoplasia, corneal pannus, cataracts, and glaucoma. <i>PAX6</i>-negative aniridia has been described in some cases, suggesting alternative genetic mechanisms. Additionally, a minority of patients with aniridia exhibit ptosis. We present a unique case of a 50-year-old woman with NF1, exhibiting bilateral congenital aniridia and ptosis, without <i>PAX6</i> mutations.</p><p><strong>Case presentation: </strong>A 50-year-old woman diagnosed with NF1 presented with bilateral congenital ptosis and aniridia. Genetic analysis confirmed the presence of the <i>NF1</i> c.4537C>T variant but was negative for <i>PAX6</i> mutations. Ophthalmological examination revealed total aniridia, cataract, ptosis, and pendular nystagmus. The patient underwent levator muscle resection for ptosis correction and cataract extraction with implantation of an intraocular lens with an iris prosthesis. Histopathological analysis of the levator muscle showed atrophic changes in the absence of neurofibromatous infiltration.</p><p><strong>Conclusion: </strong>This case represents the first documented instance of bilateral congenital aniridia in a patient with NF1. The absence of <i>PAX6</i> mutations suggests an alternative genetic mechanism or a novel NF1 phenotype. This highlights the importance of thorough ophthalmologic and genetic evaluation in NF1 patients, integrating a multidisciplinary approach to identify atypical phenotypic associations and ensure optimal management.</p>","PeriodicalId":9635,"journal":{"name":"Case Reports in Ophthalmology","volume":"16 1","pages":"461-467"},"PeriodicalIF":0.6000,"publicationDate":"2025-05-27","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12185060/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Case Reports in Ophthalmology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1159/000546420","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"Q4","JCRName":"OPHTHALMOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Introduction: Neurofibromatosis type 1 (NF1) is a genetic disorder caused by mutations in the NF1 gene on chromosome 17q11.2. The main ocular manifestations include Lisch nodules, optic pathway gliomas, and plexiform neurofibromas, all of which can potentially impair visual function. Despite the numerous documented ocular manifestations of NF1, congenital aniridia has never been previously reported. Aniridia is a rare congenital disorder primarily associated with mutations in the PAX6 gene, leading to iris hypoplasia, corneal pannus, cataracts, and glaucoma. PAX6-negative aniridia has been described in some cases, suggesting alternative genetic mechanisms. Additionally, a minority of patients with aniridia exhibit ptosis. We present a unique case of a 50-year-old woman with NF1, exhibiting bilateral congenital aniridia and ptosis, without PAX6 mutations.
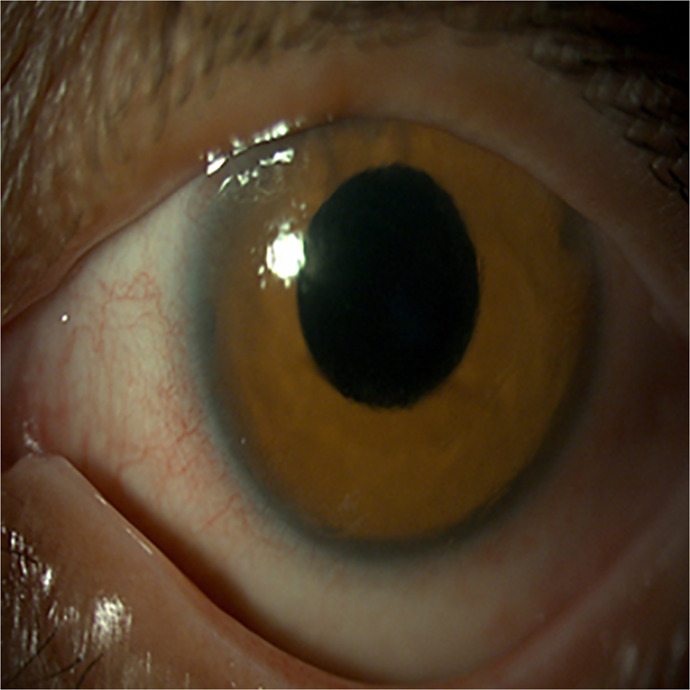
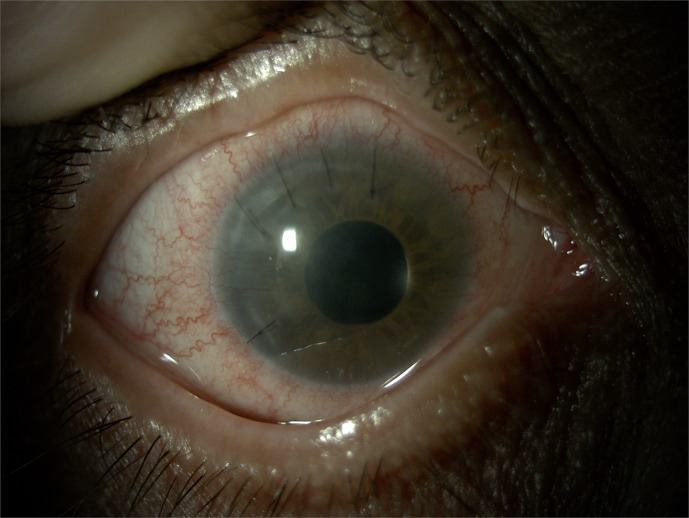
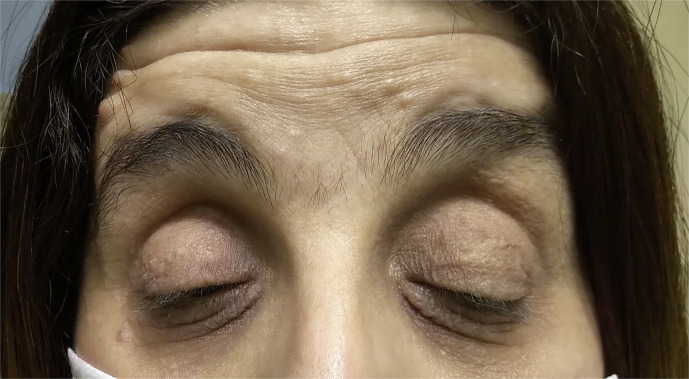
Case presentation: A 50-year-old woman diagnosed with NF1 presented with bilateral congenital ptosis and aniridia. Genetic analysis confirmed the presence of the NF1 c.4537C>T variant but was negative for PAX6 mutations. Ophthalmological examination revealed total aniridia, cataract, ptosis, and pendular nystagmus. The patient underwent levator muscle resection for ptosis correction and cataract extraction with implantation of an intraocular lens with an iris prosthesis. Histopathological analysis of the levator muscle showed atrophic changes in the absence of neurofibromatous infiltration.
Conclusion: This case represents the first documented instance of bilateral congenital aniridia in a patient with NF1. The absence of PAX6 mutations suggests an alternative genetic mechanism or a novel NF1 phenotype. This highlights the importance of thorough ophthalmologic and genetic evaluation in NF1 patients, integrating a multidisciplinary approach to identify atypical phenotypic associations and ensure optimal management.
期刊介绍:
This peer-reviewed online-only journal publishes original case reports covering the entire spectrum of ophthalmology, including prevention, diagnosis, treatment, toxicities of therapy, supportive care, quality-of-life, and survivorship issues. The submission of negative results is strongly encouraged. The journal will also accept case reports dealing with the use of novel technologies, both in the arena of diagnosis and treatment. Supplementary material is welcomed. The intent of the journal is to provide clinicians and researchers with a tool to disseminate their personal experiences to a wider public as well as to review interesting cases encountered by colleagues all over the world. Universally used terms can be searched across the entire growing collection of case reports, further facilitating the retrieval of specific information. Following the open access principle, the entire contents can be retrieved at no charge, guaranteeing easy access to this valuable source of anecdotal information at all times.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


