Epigenetic modifications are associated with mRNA and cytokine expression changes in chronic rhinosinusitis: a multiomics study from the United States.
Devyani Lal, Tripti Brar, Chantal McCabe, Erik Jessen, Nitish Kumar, Pedro Lança Gomes, Michael J Marino, Amar Miglani, Hirohito Kita
{"title":"Epigenetic modifications are associated with mRNA and cytokine expression changes in chronic rhinosinusitis: a multiomics study from the United States.","authors":"Devyani Lal, Tripti Brar, Chantal McCabe, Erik Jessen, Nitish Kumar, Pedro Lança Gomes, Michael J Marino, Amar Miglani, Hirohito Kita","doi":"10.3389/falgy.2025.1606255","DOIUrl":null,"url":null,"abstract":"<p><strong>Objectives/hypothesis: </strong>Chronic rhinosinusitis (CRS) may be triggered by environmental insults. We hypothesized that CRS results from epigenetic modifications of host DNA from external insults, leading to downstream RNA/DNA gene expression changes and immuno-mechanical disruptions. We therefore performed a multi-omics study integrating epigenetic (DNA methylation), transcriptomic (mRNA), and proteomic (cytokine) data of CRS sinonasal tissue to visualize interactions amongst these modalities to study our hypothesis.</p><p><strong>Methods: </strong>Sinonasal tissue was collected from 14 prospectively enrolled CRS and control subjects. Cytokine, mRNA transcriptome, and DNA methylome analysis were performed. Multi-omics analysis via joint dimensional reduction (JDR) was conducted.</p><p><strong>Results: </strong>Multi-omics unsupervised clustering separated subjects into two distinct groups: one cluster of 9 CRS subjects and another with 3 controls and 2 non-eosinophilic CRSsNP subjects. DNA methylation, followed by mRNA expression, contributed most to cluster assignment. DNA methylation was the most significant data modality contributing to total variance on JDR. Cytokines critical in CRS (IL-5, IL-13, IL-10, IFN<i>γ</i>, IL-6) associated with hundreds of differentially methylated regions (DMRs) and mRNA. On conjoint analyses, common upstream DMRs and mRNAs were linked to cytokines IL-5 and IL-13, cytokines IL-10 and IFN<i>γ</i>, and cytokines IFN<i>γ</i> and IL-6, respectively.</p><p><strong>Conclusions: </strong>Our results support the hypothesis that environmental insults may be significant drivers of CRS pathogenesis through epigenetic mechanisms that result in dysregulated mRNA transcription and cytokine expression. The most novel part of this study is our multi-omics approach that used integration of epigenetic (DNA methylation), transcriptomic (mRNA), and proteomic (cytokine) data to uncover insights into CRS pathogenesis; this is the first of its kind in CRS etiopathogenesis. The multi-omics analysis clearly separated clusters of control and CRS subjects, demonstrating its validity in future research. The study also identified interactions of methylated DNA, mRNA, and cytokines in CRS pathogenesis, highlighting novel molecules and pathways that may be potential therapeutic targets.</p>","PeriodicalId":73062,"journal":{"name":"Frontiers in allergy","volume":"6 ","pages":"1606255"},"PeriodicalIF":3.1000,"publicationDate":"2025-06-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12176764/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Frontiers in allergy","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3389/falgy.2025.1606255","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"ALLERGY","Score":null,"Total":0}
引用次数: 0
Abstract
Objectives/hypothesis: Chronic rhinosinusitis (CRS) may be triggered by environmental insults. We hypothesized that CRS results from epigenetic modifications of host DNA from external insults, leading to downstream RNA/DNA gene expression changes and immuno-mechanical disruptions. We therefore performed a multi-omics study integrating epigenetic (DNA methylation), transcriptomic (mRNA), and proteomic (cytokine) data of CRS sinonasal tissue to visualize interactions amongst these modalities to study our hypothesis.
Methods: Sinonasal tissue was collected from 14 prospectively enrolled CRS and control subjects. Cytokine, mRNA transcriptome, and DNA methylome analysis were performed. Multi-omics analysis via joint dimensional reduction (JDR) was conducted.
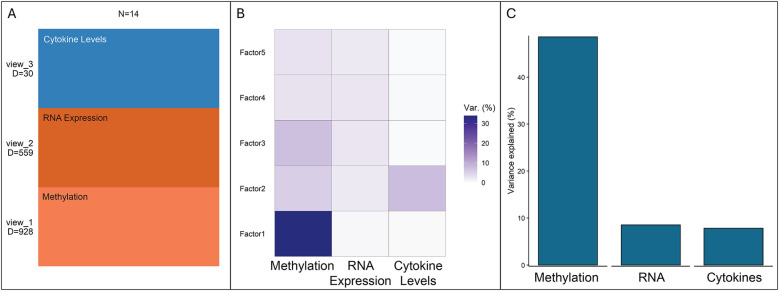
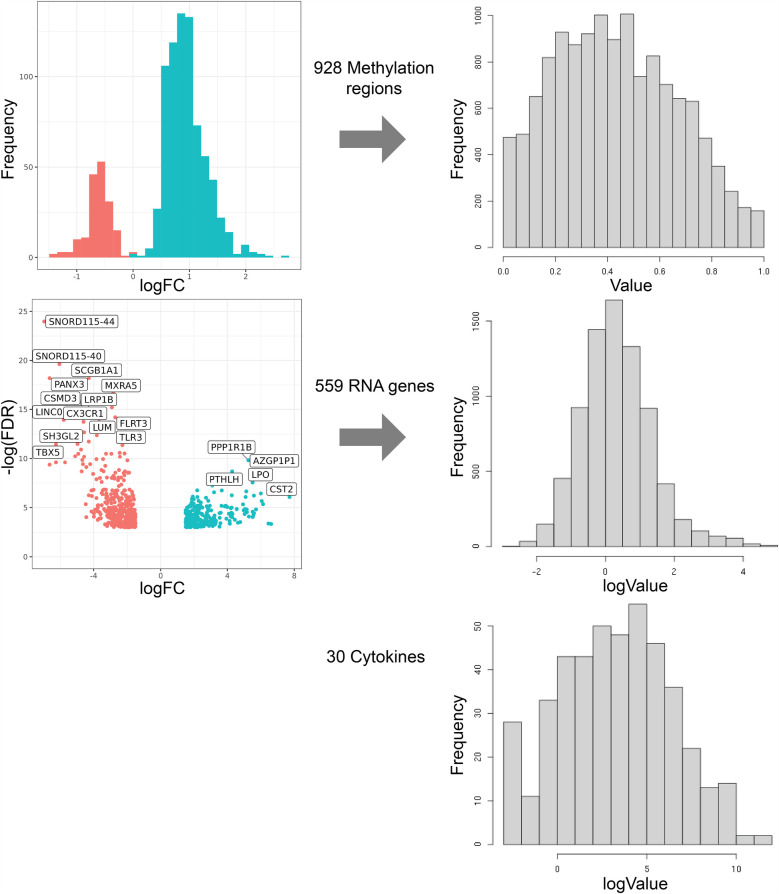
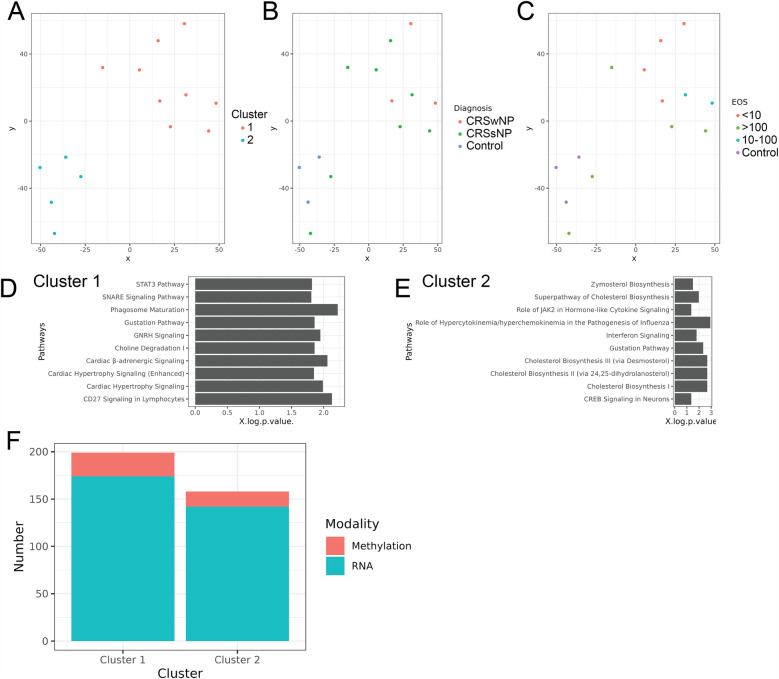
Results: Multi-omics unsupervised clustering separated subjects into two distinct groups: one cluster of 9 CRS subjects and another with 3 controls and 2 non-eosinophilic CRSsNP subjects. DNA methylation, followed by mRNA expression, contributed most to cluster assignment. DNA methylation was the most significant data modality contributing to total variance on JDR. Cytokines critical in CRS (IL-5, IL-13, IL-10, IFNγ, IL-6) associated with hundreds of differentially methylated regions (DMRs) and mRNA. On conjoint analyses, common upstream DMRs and mRNAs were linked to cytokines IL-5 and IL-13, cytokines IL-10 and IFNγ, and cytokines IFNγ and IL-6, respectively.
Conclusions: Our results support the hypothesis that environmental insults may be significant drivers of CRS pathogenesis through epigenetic mechanisms that result in dysregulated mRNA transcription and cytokine expression. The most novel part of this study is our multi-omics approach that used integration of epigenetic (DNA methylation), transcriptomic (mRNA), and proteomic (cytokine) data to uncover insights into CRS pathogenesis; this is the first of its kind in CRS etiopathogenesis. The multi-omics analysis clearly separated clusters of control and CRS subjects, demonstrating its validity in future research. The study also identified interactions of methylated DNA, mRNA, and cytokines in CRS pathogenesis, highlighting novel molecules and pathways that may be potential therapeutic targets.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


