Xu Zhang, Lin Tao, Davoud Dastan, Hongwei Zhang and Baochang Gao
{"title":"Tuning the electrochemical stability of carbon based single-atom structures via doping: trade-off between electrosorption/leaching behavior†","authors":"Xu Zhang, Lin Tao, Davoud Dastan, Hongwei Zhang and Baochang Gao","doi":"10.1039/D5TA03307A","DOIUrl":null,"url":null,"abstract":"<p >The performance of single-atom catalysts in electrocatalytic processes can be effectively enhanced through the doping of tailored asymmetric coordination environments. However, understanding the electrochemical stability of doped single-atom structures (SAS) under operating conditions remains challenging. In this study, density functional theory (DFT) and <em>ab initio</em> molecular dynamics (AIMD) simulations are employed to elucidate the combined effects of proton–electron adsorption and Cu leaching from the Cu–N<small><sub>4</sub></small> structure. By considering 6 thermodynamically and kinetically stable heteroatom-doped CuN<small><sub>3</sub></small>X structures, the relationship between the proton–electron adsorption energy barriers and Cu leaching energy barriers for 96 proton adsorption configurations is explored. A trade-off between these two factors leads to the identification of the CuN<small><sub>3</sub></small>B structure as the most stable. Surface phase diagrams indicate that B doping effectively suppresses Cu leaching, while S doping exacerbates it. Electronic structure analysis further highlights that B doping enhances the hybridization coincidence of Cu–N orbitals, thereby strengthening the Cu–N bond, reducing proton adsorption on N, and ultimately stabilizing the Cu single-atom structure. Overall, this study investigates the electrochemical stability of Cu SAS and their underlying mechanisms, offering new insights into the electrochemical stability of SAS.</p>","PeriodicalId":82,"journal":{"name":"Journal of Materials Chemistry A","volume":" 29","pages":" 23715-23723"},"PeriodicalIF":9.5000,"publicationDate":"2025-06-20","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Materials Chemistry A","FirstCategoryId":"88","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2025/ta/d5ta03307a","RegionNum":2,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
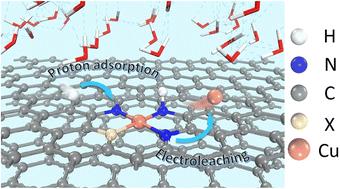
The performance of single-atom catalysts in electrocatalytic processes can be effectively enhanced through the doping of tailored asymmetric coordination environments. However, understanding the electrochemical stability of doped single-atom structures (SAS) under operating conditions remains challenging. In this study, density functional theory (DFT) and ab initio molecular dynamics (AIMD) simulations are employed to elucidate the combined effects of proton–electron adsorption and Cu leaching from the Cu–N4 structure. By considering 6 thermodynamically and kinetically stable heteroatom-doped CuN3X structures, the relationship between the proton–electron adsorption energy barriers and Cu leaching energy barriers for 96 proton adsorption configurations is explored. A trade-off between these two factors leads to the identification of the CuN3B structure as the most stable. Surface phase diagrams indicate that B doping effectively suppresses Cu leaching, while S doping exacerbates it. Electronic structure analysis further highlights that B doping enhances the hybridization coincidence of Cu–N orbitals, thereby strengthening the Cu–N bond, reducing proton adsorption on N, and ultimately stabilizing the Cu single-atom structure. Overall, this study investigates the electrochemical stability of Cu SAS and their underlying mechanisms, offering new insights into the electrochemical stability of SAS.
期刊介绍:
The Journal of Materials Chemistry A, B & C covers a wide range of high-quality studies in the field of materials chemistry, with each section focusing on specific applications of the materials studied. Journal of Materials Chemistry A emphasizes applications in energy and sustainability, including topics such as artificial photosynthesis, batteries, and fuel cells. Journal of Materials Chemistry B focuses on applications in biology and medicine, while Journal of Materials Chemistry C covers applications in optical, magnetic, and electronic devices. Example topic areas within the scope of Journal of Materials Chemistry A include catalysis, green/sustainable materials, sensors, and water treatment, among others.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


