Felipe Augusto Azevedo Leão, Leticia Ferreira Gontijo Silveira, Rodrigo Rezende Arantes, Milena Maria Moreira Guimarães
{"title":"Clinical features and genetic analysis of a Brazilian patient with sitosterolemia: a case report.","authors":"Felipe Augusto Azevedo Leão, Leticia Ferreira Gontijo Silveira, Rodrigo Rezende Arantes, Milena Maria Moreira Guimarães","doi":"10.20945/2359-4292-2024-0326","DOIUrl":null,"url":null,"abstract":"<p><p>Sitosterolemia is a rare genetic lipid disorder caused by mutations in the ABCG5/ABCG8, genes. It is characterized by plasmatic plant sterols accumulation, formation of tendon and tuberous xanthomas and early onset coronary artery disease. The differential diagnosis with other congenital dyslipidemias presents significant challenges. We describe a case of a male patient who presented with hypercholesterolemia and tendinous xantomas from the age of 5. The patient was born to consanguineous parents, with no family history of hypercholesterolemia. With the initial hypothesis of cerebrotendinous xanthomatosis, he was treated with chenodeoxycholic acid, which yielded no improvement. Over time, he developed persistent thrombocytopenia and arthralgia, and experienced an acute myocardial infarction at the age of 27. Genetic analysis revealed the previously known p.Trp361*mutation in homozygosity in the ABCG8 gene and was negative for CYP27A1 variants, associated with cerebrotendinous xanthomatosis. The subsequent introduction of a diet with vegetable fats restriction and administration of ezetimibe resulted in an excellent response. The diagnosis of congenital hypercholesterolemia is challenging due to the low prevalence and heterogenous presentation of the condition. This case underscores the importance of clinical suspicion and the confirmation of the molecular diagnosis for a precise therapeutic management.</p>","PeriodicalId":54303,"journal":{"name":"Archives of Endocrinology Metabolism","volume":"69 3","pages":"e240326"},"PeriodicalIF":2.3000,"publicationDate":"2025-06-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12176087/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Archives of Endocrinology Metabolism","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.20945/2359-4292-2024-0326","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
引用次数: 0
Abstract
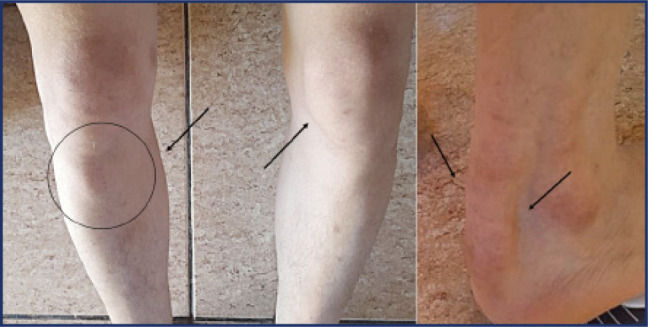
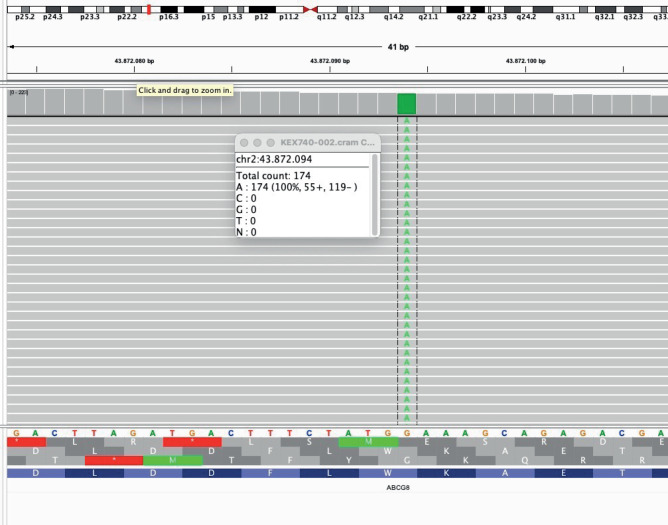
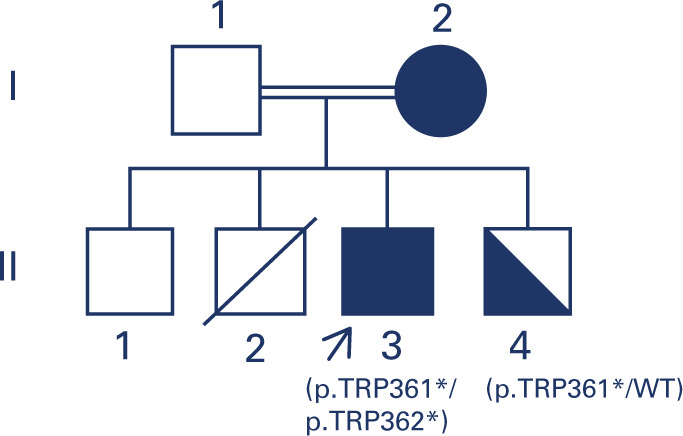
Sitosterolemia is a rare genetic lipid disorder caused by mutations in the ABCG5/ABCG8, genes. It is characterized by plasmatic plant sterols accumulation, formation of tendon and tuberous xanthomas and early onset coronary artery disease. The differential diagnosis with other congenital dyslipidemias presents significant challenges. We describe a case of a male patient who presented with hypercholesterolemia and tendinous xantomas from the age of 5. The patient was born to consanguineous parents, with no family history of hypercholesterolemia. With the initial hypothesis of cerebrotendinous xanthomatosis, he was treated with chenodeoxycholic acid, which yielded no improvement. Over time, he developed persistent thrombocytopenia and arthralgia, and experienced an acute myocardial infarction at the age of 27. Genetic analysis revealed the previously known p.Trp361*mutation in homozygosity in the ABCG8 gene and was negative for CYP27A1 variants, associated with cerebrotendinous xanthomatosis. The subsequent introduction of a diet with vegetable fats restriction and administration of ezetimibe resulted in an excellent response. The diagnosis of congenital hypercholesterolemia is challenging due to the low prevalence and heterogenous presentation of the condition. This case underscores the importance of clinical suspicion and the confirmation of the molecular diagnosis for a precise therapeutic management.
期刊介绍:
The Archives of Endocrinology and Metabolism - AE&M – is the official journal of the Brazilian Society of Endocrinology and Metabolism - SBEM, which is affiliated with the Brazilian Medical Association.
Edited since 1951, the AE&M aims at publishing articles on scientific themes in the basic translational and clinical area of Endocrinology and Metabolism. The printed version AE&M is published in 6 issues/year. The full electronic issue is open access in the SciELO - Scientific Electronic Library Online e at the AE&M site: www.aem-sbem.com.
From volume 59 on, the name was changed to Archives of Endocrinology and Metabolism, and it became mandatory for manuscripts to be submitted in English for the online issue. However, for the printed issue it is still optional for the articles to be sent in English or Portuguese.
The journal is published six times a year, with one issue every two months.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


