Isabel Rancu, Benjamin Sobkowiak, Joshua L Warren, Nelly Ciobanu, Alexandru Codreanu, Valeriu Crudu, Caroline Colijn, Ted Cohen, Melanie H Chitwood
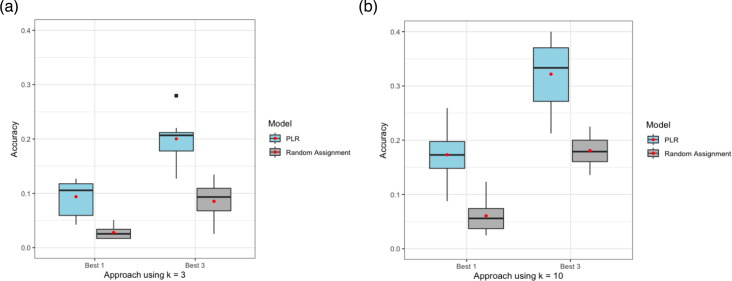
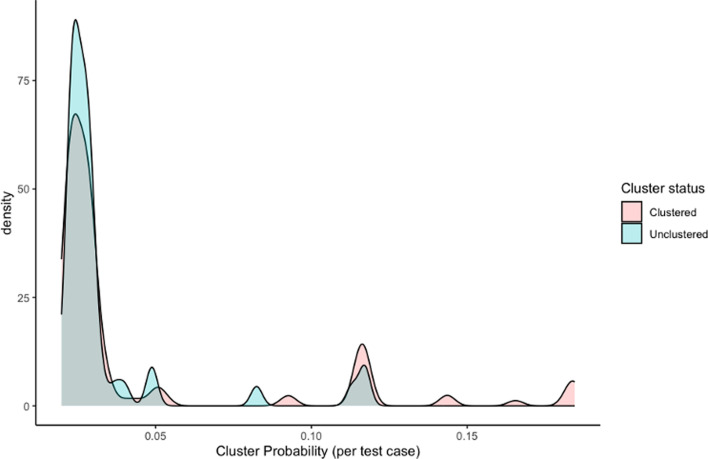
{"title":"Classification of unsequenced Mycobacterium tuberculosis strains in a high-burden setting using a pairwise logistic regression approach.","authors":"Isabel Rancu, Benjamin Sobkowiak, Joshua L Warren, Nelly Ciobanu, Alexandru Codreanu, Valeriu Crudu, Caroline Colijn, Ted Cohen, Melanie H Chitwood","doi":"10.1099/acmi.0.000964.v3","DOIUrl":null,"url":null,"abstract":"<p><p>Over the past three decades, molecular epidemiological studies have provided new opportunities to investigate the transmission dynamics of <i>Mycobacterium tuberculosis</i>. In most studies, a sizable fraction of individuals with notified tuberculosis cannot be included, either because they do not have culture-positive disease (and thus do not have specimens available for molecular typing) or because resources for conducting sequencing are limited. A recent study introduced a regression-based approach for inferring the membership of unsequenced tuberculosis cases in transmission clusters based on host demographic and epidemiological data. This method was able to identify the most likely cluster to which an unsequenced strain belonged with an accuracy of 35%, although this was in a low-burden setting where a large fraction of cases occurred among foreign-born migrants. Here, we apply a similar model to <i>M. tuberculosis</i> whole-genome sequencing data from the Republic of Moldova, a setting of relatively high local transmission. Using a maximum cluster span of ~40 single nucleotide polymorphisms (SNPs) and a cluster size cutoff of <i>n</i>≥10, we could best predict the specific cluster to which each clustered case was most likely to be a member with an accuracy of 17.2 %. In sensitivity analyses, we found that a more restrictive (~20 SNPs threshold) or permissive (~80 SNPs) threshold did not improve performance. We found that increasing the minimum cluster size improved prediction accuracy. These findings highlight the challenges of transmission inference in high-burden settings like Moldova.</p>","PeriodicalId":94366,"journal":{"name":"Access microbiology","volume":"7 5","pages":""},"PeriodicalIF":0.0000,"publicationDate":"2025-05-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12163731/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Access microbiology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1099/acmi.0.000964.v3","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
Over the past three decades, molecular epidemiological studies have provided new opportunities to investigate the transmission dynamics of Mycobacterium tuberculosis. In most studies, a sizable fraction of individuals with notified tuberculosis cannot be included, either because they do not have culture-positive disease (and thus do not have specimens available for molecular typing) or because resources for conducting sequencing are limited. A recent study introduced a regression-based approach for inferring the membership of unsequenced tuberculosis cases in transmission clusters based on host demographic and epidemiological data. This method was able to identify the most likely cluster to which an unsequenced strain belonged with an accuracy of 35%, although this was in a low-burden setting where a large fraction of cases occurred among foreign-born migrants. Here, we apply a similar model to M. tuberculosis whole-genome sequencing data from the Republic of Moldova, a setting of relatively high local transmission. Using a maximum cluster span of ~40 single nucleotide polymorphisms (SNPs) and a cluster size cutoff of n≥10, we could best predict the specific cluster to which each clustered case was most likely to be a member with an accuracy of 17.2 %. In sensitivity analyses, we found that a more restrictive (~20 SNPs threshold) or permissive (~80 SNPs) threshold did not improve performance. We found that increasing the minimum cluster size improved prediction accuracy. These findings highlight the challenges of transmission inference in high-burden settings like Moldova.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


