Mariusz Berdyński, Krzysztof Safranow, Peter M. Andersen, Cezary Żekanowski
{"title":"Phenotypic Characterization of ALS-Causing SOD1 Mutations Affecting Polypeptide Length","authors":"Mariusz Berdyński, Krzysztof Safranow, Peter M. Andersen, Cezary Żekanowski","doi":"10.1155/humu/9792233","DOIUrl":null,"url":null,"abstract":"<p><b>Background:</b> Some 234 mutations in the small <i>SOD1</i> gene have been reported to cause amyotrophic lateral sclerosis. However, the pathogenic mechanisms, particularly of those mutations affecting polypeptide length, are contested. It is presently unknown whether all reported nonsense mutations in SOD1 are causative for ALS. The emergence of promising new anti-SOD1 drugs has made it imperative to gain further insight into clinical–genetic aspects of ALS for deciding which patients to treat in clinical practice and include in drug trials.</p><p><b>Objective:</b> This study is aimed at comprehensively analyzing the clinical phenotypes associated with ALS-causing SOD1 mutations that alter the polypeptide length. The specific focus is on the age at which symptoms manifest and the survival duration.</p><p><b>Methods:</b> Data were collected from web databases, published reports, conference presentations, and personal communications up to November 2023. The clinical endpoints, including age at symptom onset and age at death, were subjected to survival analysis. Comparative analyses were performed between frameshift and nonframeshift variants.</p><p><b>Results:</b> A cohort of 146 ALS patients harboring 38 different nonmissense <i>SOD1</i> variants was analyzed. The mean age of disease onset was 46.9 years, with a mean survival duration of 49 months. Significant heterogeneity was observed in clinical outcomes, with earlier disease onset and reduced survival associated with specific mutations. Notably, frameshift mutations proximal to the N-terminus showed a higher risk of early ALS onset compared to more distal mutations.</p><p><b>Conclusions:</b> The clinical phenotypes of ALS patients with nonmissense SOD1 mutations are highly variable and dependent on the specific mutation. These findings underscore the necessity of including diverse <i>SOD1</i> mutation carriers in therapeutic trials and suggest that both loss-of-function and gain-of-function mechanisms may contribute to ALS pathology.</p>","PeriodicalId":13061,"journal":{"name":"Human Mutation","volume":"2025 1","pages":""},"PeriodicalIF":3.7000,"publicationDate":"2025-06-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1155/humu/9792233","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Human Mutation","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1155/humu/9792233","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Some 234 mutations in the small SOD1 gene have been reported to cause amyotrophic lateral sclerosis. However, the pathogenic mechanisms, particularly of those mutations affecting polypeptide length, are contested. It is presently unknown whether all reported nonsense mutations in SOD1 are causative for ALS. The emergence of promising new anti-SOD1 drugs has made it imperative to gain further insight into clinical–genetic aspects of ALS for deciding which patients to treat in clinical practice and include in drug trials.
Objective: This study is aimed at comprehensively analyzing the clinical phenotypes associated with ALS-causing SOD1 mutations that alter the polypeptide length. The specific focus is on the age at which symptoms manifest and the survival duration.
Methods: Data were collected from web databases, published reports, conference presentations, and personal communications up to November 2023. The clinical endpoints, including age at symptom onset and age at death, were subjected to survival analysis. Comparative analyses were performed between frameshift and nonframeshift variants.
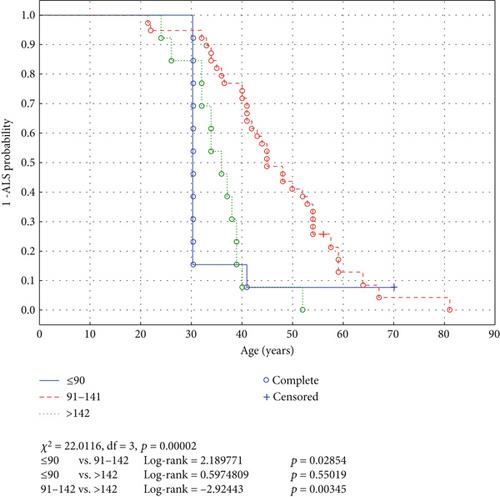
Results: A cohort of 146 ALS patients harboring 38 different nonmissense SOD1 variants was analyzed. The mean age of disease onset was 46.9 years, with a mean survival duration of 49 months. Significant heterogeneity was observed in clinical outcomes, with earlier disease onset and reduced survival associated with specific mutations. Notably, frameshift mutations proximal to the N-terminus showed a higher risk of early ALS onset compared to more distal mutations.
Conclusions: The clinical phenotypes of ALS patients with nonmissense SOD1 mutations are highly variable and dependent on the specific mutation. These findings underscore the necessity of including diverse SOD1 mutation carriers in therapeutic trials and suggest that both loss-of-function and gain-of-function mechanisms may contribute to ALS pathology.
期刊介绍:
Human Mutation is a peer-reviewed journal that offers publication of original Research Articles, Methods, Mutation Updates, Reviews, Database Articles, Rapid Communications, and Letters on broad aspects of mutation research in humans. Reports of novel DNA variations and their phenotypic consequences, reports of SNPs demonstrated as valuable for genomic analysis, descriptions of new molecular detection methods, and novel approaches to clinical diagnosis are welcomed. Novel reports of gene organization at the genomic level, reported in the context of mutation investigation, may be considered. The journal provides a unique forum for the exchange of ideas, methods, and applications of interest to molecular, human, and medical geneticists in academic, industrial, and clinical research settings worldwide.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


