RIPTACs for Precision Cancer Therapy: A Novel Modality with the Inspiration of HLD-0915 as the First Candidate in Clinical Trials
IF 6.8
1区 医学
Q1 CHEMISTRY, MEDICINAL
引用次数: 0
Abstract
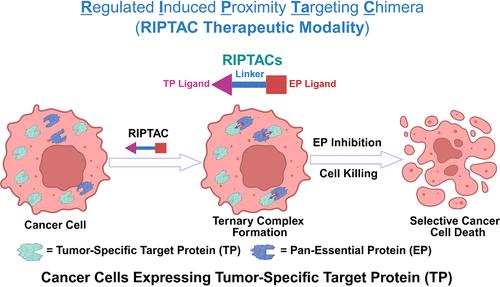
Chemically induced proximity-based approaches are promising alternatives to small molecule inhibitors, attracting increasing attention in the drug discovery field. Small molecule chemical inducers of proximity (CIPs) can induce molecular proximity of two proteins in living cells. CIPs are developed to recapitulate diverse biological processes in living cells and organisms, including transcription, post-translational modification, and signal transduction. (1) Based on an “event-driven” model instead of “occupancy-driven” model by small molecule inhibitors, CIPs are able to drive a cellular event with a substoichiometric binding of proteins. (2) As an important member of CIPs, proteolysis-targeting chimeras (PROTACs) catalytically induce the proximity between cellular protein-of-interest (POI) and the E3 ligase, causing the polyubiquitination and degradation of the POI. (3) Over the past two decades, the emergence and prosperity of PROTAC technology opened new avenues for drug discovery and development with about 20 PROTAC molecules in human clinical trials. (4−11) Inspired by the great success of PROTACs, (12) several new CIPs beyond degradation have been developed, (13,14) such as regulated induced proximity targeting chimeras (RIPTACs), (15,16) deubiquitinase-targeting chimeras (DUBTACs), (17−20) enhancement-targeting chimeras (ENTACs), (21) RESTORACs, (22) phosphorylation-inducing chimeras (PHICs), (23) phosphatase-recruiting chimeras (PhoRCs), (24,25) phosphorylation-targeting chimeras (PhosTACs), (26) dephosphorylation-targeting chimeras (DEPTACs), (27) acetylation tagging system (AceTAG), (28,29) and transcriptional/epigenetic CIPs (TCIPs). (30−32) RIPTACs are emerging as a new therapeutic modality for various cancers (Figure 1). Like PROTACs, RIPTACs are also a class of heterobifunctional molecules, comprising a ligand for the tumor-specific target protein (TP) that is selectively expressed in cancer cells, a ligand for the pan-Essential Protein (EP) that is required for cell survival, and a linker connecting these two components. RIPTACs selectively kill cancer cells expressing tumor-specific TP while sparing non-TP expressing healthy cells. RIPTACs selectively accumulate in the cancer cells expressing tumor-specific TP and form a stable ternary complex between the TP and EP (TP:RIPTAC:EP). The stable ternary complex induces or enhances the protein–protein interactions (PPIs) between the TP and EP, abrogating the EP function and thus causing selective cancer cell death. RIPTACs role as a class of bivalent molecular glue inhibitors by bringing the TP and EP into proximity in cancer cells, delivering a “hold and kill” blow. Figure 1. Schematic diagram of RIPTAC therapeutic modality. RIPTACs are a novel class of heterobifunctional molecules, comprising a TP ligand, an EP ligand, and a connecting linker. RIPTACs selectively accumulate in cancer cells expressing tumor-specific TP, form a stable ternary complex with the TP and EP (TP:RIPTAC:EP), and abrogate the EP function, leading to selective cancer cell death while sparing healthy cells. RIPTACs can induce neo-PPIs or enhance existing PPIs, acting as bivalent molecular glue inhibitors with a “hold and kill” mechanism. RIPTACs have the potential to mitigate the on-target off-tumor toxicity caused by the EPs. As a paradigm-shifting drug discovery approach, RIPTACs are attracting more and more attention from both academic and industrial settings. Recently, Halda Therapeutics, founded by Crews and co-workers, announced that the first patient with metastatic castration-resistant prostate cancer (mCRPC) is administrated with HLD-0915, an androgen receptor (AR)-targeting RIPTAC, in a Phase 1/2 clinical trial (NCT06800313). (33) The purpose of this first-in-human clinical trial is to evaluate the safety, tolerability, pharmacokinetics (PK), pharmacodynamics (PD), and antitumor activity of orally dosed single-agent HLD-0915. (33,34) This study includes an initial Phase 1 and a subsequent Phase 2 clinical trials. The Phase 1 clinical trial is a dose escalation study to determine the maximum tolerated dose (MTD) and/or recommended dose(s) of HLD-0915 as a monotherapy. The Phase 2 expansion cohort is aimed to further evaluate the efficacy and safety of HLD-0915. The Phase 1/2 clinical study plans to enroll up to 80 mCRPC patients. HLD-0915 is a heterobifunctional molecule, comprising an AR ligand as the TP, an EP ligand, and a connecting linker. So far, the EP and chemical structure of HLD-0915 have not been disclosed. HLD-0915 is orally effective in preclinical prostate cancer models, including drug resistant models, resulting in significant tumor shrinkage and prostate-specific antigen (PSA) reduction, while showing a favorable therapeutic index. Prostate cancer is the second most common malignancy and the fifth leading cause of cancer death among men worldwide, with about 1.5 million new cases and 397,000 deaths in 2022. (35) Almost all prostate cancer patients develop drug resistance to AR signaling inhibitors, which is driven by many heterogeneous bypass resistance mechanisms, including genetic mutations in AR and increased expression of AR levels. The AR gene, which codes for AR, or the upstream enhancer region of DNA are aberrantly amplified in more than 80% mCRPC patients. (36) Clinical needs remain unmet for prostate cancer patients, especially those with advanced, drug resistant, and the most lethal type. Thus, novel therapies are urgently needed to tackle such challenging diseases. AR-targeting RIPTACs (e.g., HLD-0915) with a unique mechanism of action are emerging as a promising alternative precision cancer therapy for mCRPC. To develop novel therapeutics for treating prostate cancer resistant to AR antagonists, such as enzalutamide, researchers at Halda Therapeutic designed and synthesized a series of AR-targeting heterobifunctional molecules. Excitingly, two compounds, H001 and H003, were demonstrated to be promising candidates for treating prostate cancer. (33) H001 and H003 are bona fide RIPTACs, acting through a chemically induced proximity mechanism. H001 and H003 form a stable ternary complex with the TP (AR) and EP, which is revealed by the X-ray cocrystal structure of the AR:RIPTAC:EP ternary complex, but the PDB code has not been disclosed. The determination of the X-ray crystal structure of the AR:RIPTAC:EP ternary complex enables structure-based drug design (SBDD) of AR-targeting RIPTACs. Both H001 and H003 form an intracellular ternary complex with AR and the EP in VcaP cells with EC50 values of 24 nM and 9 nM, respectively. However, neither the AR ligand nor the EP ligand alone induces the formation of the intracellular ternary complex, indicating that both the AR ligand and the EP ligand within RIPTACs are involved in forming the ternary complex. Additionally, AR-targeting RIPTACs exhibit positive co-operativity in AR binding and TP binding, contributing to forming a more stable ternary complex. H001 and H003 display potent antiproliferation activity (H001: IC50 < 10 nM; H003: IC50 < 1 nM) and EP inhibition in Trex293 cells expressing doxycycline-inducible AR. Compared to the parental 22RV1 cells expressing low levels of FL-AR, H001 and H003 significantly induce cell apoptosis in 22RV1 cells overexpressing FL-AR (H001: EC50 = 113 nM; H003: EC50 = 48.5 nM), as determined by the Caspase 3/7 GIo assay. 22RV1/AR cells overexpressing FL-AR are derived from the parental AR-V7+ cells, an Enzalutamide-resistant cell line. H001 induces the formation of AR:RIPTAC:EP ternary complex in Trex293 cells expressing clinically relevant AR mutants (ARMut) induced by doxycycline. Doxycycline-induced ARMut leads to the substitution of lysine residue at position 702 by histidine (L702H), histidine residue at position 875 by tyrosine (H875Y), and threonine residue at position 878 by alanine (T878A). H001 and H003 (30 mg/kg, p.o., QD) display superior oral in vivo efficacy to Enzalutamide, an AR signaling inhibitor, in an AR-amplified (ARamp), V7+ VCaP prostate tumor model in castrated mice. H001 and H003 cause tumor regression in a VCaP tumor model in castrated mice, an Enzalutamide-insensitive prostate cancer model. Moreover, H001 and H003 significantly reduce the plasma PSA. Furthermore, H001 and H003 can form a ternary complex with AR and EP in the tumor tissue and resulted in EP inhibition, transforming into robust in vivo efficacy. Predosing with Enzalutamide that occupies the AR ligand binding domain (AR-LBD) competes off AR binding with H001, leading to significant attenuation in the ternary complex (AR:H001:EP) formation and EP inhibition in a VCaP tumor model in castrated mice. This finding indicates that the in vivo pharmacodynamic (PD) modulation of H001 is dependent on AR binding. H001 induces “BRCAness” in both in vitro and in vivo VCaP models. H001 dramatically suppresses the expression of breast cancer gene 1 (BRCA1), breast cancer gene 2 (BRCA2), and related homologous recombination repair (HRR) genes, such as RAD51-associated protein 1 (RAD51AP1), RecQ-mediated genome instability protein 2 (RMI2), and RAD54-like protein (RAD54L), in VCaP cells in a dose-dependent manner. Moreover, H001 (30 mg/kg) is orally effective for inhibiting the expression of these HRR genes in a castrate VCaP tumor model. PARP inhibitors can cause synthetic lethality in prostate cancer cells bearing BRCA1/2 deficiency. Therefore, H001 in combination with PARP inhibitors may exert synergetic effect for treating prostate cancers, showing the potential to overcome drug-resistance to PARP inhibitors. Combination therapy studies of AR-RIPTACs and PARP inhibitors will be anticipated. RIPTACs have several unique features in comparison with existing therapeutic modalities as outlined in Table 1. First, RIPTACs exert their function independent of the TP function, which is similar to bispecific antibodies (bsAbs), antibody–drug conjugates (ADCs) and CAR-T-mAb, but different with small molecule inhibitors and protein degraders. Second, RIPTACs are effective for drug-resistant cancer cells caused by nontarget-based resistance mechanisms, which is similar to bsAbs, ADCs, and CAR-T-mAb, but different with small molecule inhibitors and protein degraders. Third, RIPTACs show potential to treat cancers by targeting tumor-driving “undruggable” oncoproteins, which is similar to protein degraders, bsAbs, ADCs, and CAR-T-mAb, but superior to small molecule inhibitors. Fourth, RIPTACs can induce the formation of TP:RIPTAC:EP ternary complex, inducing or enhancing the PPIs between the TP and EP, which is similar to protein degraders, such as molecular glue degraders (MGDs), but different with small molecule inhibitors, bsAbs, ADCs, and CAR-T-mAb. Fifth, like small molecule inhibitors and protein degraders, RIPTACs can be developed as oral dosage with a low cost of goods sold (COGS), which is not accessed by bsAbs, ADCs, and CAR-T-mAb. In addition, like small molecule inhibitors and protein degraders, RIPTACs target intracellular TPs, which is different with bsAbs, ADCs, and CAR-T-mAb relying on the antigen on the cell membrane. RIPTACs present several advantages over existing therapeutic modalities. First, RIPTACs selectively kill cancer cells while sparing normal cells, delivering a high therapeutic index. Second, RIPTACs take effect independent of the TP function, largely expanding the scope of TP. To this end, some “undruggable” targets specifically expressing in tumor cells can be used as TPs for developing RIPTACs. Third, based on the positively cooperative binding mechanism, RIPTACs do not need to have a high binding affinity with the TPs, showing the potential to overcome drug resistance caused by TP mutations. Moreover, RIPTACs also have the potential to overcome drug resistance caused by nontarget-based resistance mechanisms. Nevertheless, challenges also exist for the development of RIPTACs. Like PROTACs, RIPTACs also have a relatively large molecular weight, which is beyond the Lipinski’s “Rule of Five” (Ro5), limiting the overall druglike properties such as cell permeability, aqueous solubility, and metabolic stability. Positive clinical results of PROTACs partially attenuate such a concern through systematic and continuous structural optimizations. Moreover, the successful advancement of HLD-0915 into clinical trials inspires the endeavor of RIPTAC modality for the drug development. Given the unique molecular glue mechanism, the requirements for the ligand selection of the TP and EP, as well as the type and length of the linker are stricter for RIPTACs than PROTACs. Thus, the success rate of developing RIPTACs is lower than that of PROTACs. Few of the designed and synthesized bivalent heterobifunctional molecules as potential RIPTACs are bona fide RIPTACs, while most of them may simply act as potential dual inhibitors without inducing proximity. Furthermore, the number of TPs explored for developing RIPTACs is largely limited. Currently, only AR, ER, FKBP, and P53 Y220C proteins have been determined as suitable TPs for RIPTACs. (15,16,37,38) More diverse TPs need to be explored for developing novel RIPTACs. (38) The EPs used for RIPTACs mainly include cyclin-dependent kinases (CDKs), bromodomain and extra terminal domain (BET) proteins, and polo-like kinase 1 (PLK1). (15,16) In addition, RIPTACs are currently exploited primarily for treating cancers, while this strategy may also be expanded and applicable for other threatening diseases in the future. In conclusion, RIPTAC technology is a novel chemically induced proximity-based therapeutic strategy, opening new avenues for innovative drug discovery and development. RIPTACs with a unique selective and wide applicable cancer cell-killing mechanism have the potential to mitigate side effects including on-target off-tissue toxicity and overcome drug resistance. As an emerging technology, developing RIPTACs is still in an early stage, but inspired by the milestone of successful advancement of a first RIPTAC HLD-0915 into Phase 1/2 clinical trial for treating mCRPC (NCT06800313). (34) While we look forward to the positive clinical outcomes of these studies, RIPTACs as a novel modality may offer an unparalleled opportunity for developing precision medicines, particularly for cancer therapy. This work was partially supported by R01CA226001 and R01CA231150 grants from the National Institutes of Health, Breast Cancer Research Program (BCRP) Breakthrough Awards W81XWH-17-1-0071 and W81XWH-17-1-0072 from the Department of Defense (DoD), the John D. Stobo, M.D. Distinguished Chair Endowment, and the Edith & Robert Zinn Chair Endowment in Drug Discovery. This article references 38 other publications. This article has not yet been cited by other publications.
RIPTACs用于癌症精准治疗:HLD-0915作为临床试验第一候选药物的新模式
化学诱导的基于接近的方法是小分子抑制剂的有希望的替代品,在药物发现领域引起了越来越多的关注。小分子化学接近诱导剂(CIPs)可以在活细胞中诱导两种蛋白质的分子接近。cip的发展是为了概括活细胞和生物体中的各种生物过程,包括转录、翻译后修饰和信号转导。(1)基于“事件驱动”模型而不是小分子抑制剂的“占用驱动”模型,cip能够通过亚化学计量的蛋白质结合来驱动细胞事件。(2)蛋白水解靶向嵌合体(proteolysis-targeting chimeras, PROTACs)作为cip的重要成员,可催化诱导细胞感兴趣蛋白(POI)与E3连接酶的接近,导致POI的多泛素化和降解。(3)在过去的二十年中,PROTAC技术的出现和繁荣为药物发现和开发开辟了新的途径,大约有20个PROTAC分子进入人体临床试验。(4−11)受到PROTACs的巨大成功的启发,(12)已经开发了几种超越降解的新型cip,(13,14)如调节诱导近距离靶向嵌合体(RIPTACs),(15,16)去ubiquitinase靶向嵌合体(DUBTACs),(17−20)增强靶向嵌合体(ENTACs), (21) RESTORACs,(22)磷酸化诱导嵌合体(PHICs),(23)磷酸酶募集嵌合体(PhoRCs),(24,25)磷酸化靶向嵌合体(PhosTACs),(26)去磷酸化靶向嵌合体(DEPTACs),(27)乙酰化标记系统(AceTAG),(28,29)和转录/表观遗传cip (tcip)。(30−32)riptac正在成为各种癌症的一种新的治疗方式(图1)。与PROTACs一样,riptac也是一类异双功能分子,包括在癌细胞中选择性表达的肿瘤特异性靶蛋白(TP)的配体,细胞存活所需的泛必需蛋白(EP)的配体,以及连接这两个成分的连接子。riptac选择性地杀死表达肿瘤特异性TP的癌细胞,同时保留非TP表达的健康细胞。RIPTAC选择性地在表达肿瘤特异性TP的癌细胞中积累,并在TP和EP之间形成稳定的三元复合物(TP:RIPTAC:EP)。稳定的三元配合物诱导或增强TP和EP之间的蛋白-蛋白相互作用(PPIs),取消EP功能,从而导致选择性癌细胞死亡。RIPTACs作为一类二价分子胶抑制剂,通过将TP和EP靠近癌细胞,提供“保持和杀死”的打击。图1所示。RIPTAC治疗方式示意图。riptac是一类新型的异双功能分子,由一个TP配体、一个EP配体和一个连接体组成。RIPTAC选择性地在表达肿瘤特异性TP的癌细胞中积累,与TP和EP形成稳定的三元配合物(TP:RIPTAC:EP),并破坏EP功能,导致癌细胞选择性死亡,同时保留健康细胞。RIPTACs可以诱导新的PPIs或增强现有的PPIs,作为具有“保持和杀死”机制的二价分子胶抑制剂。riptac具有减轻EPs引起的靶外肿瘤毒性的潜力。RIPTACs作为一种范式转换的药物发现方法,越来越受到学术界和工业界的关注。最近,由Crews及其同事创立的Halda Therapeutics公司宣布,在1/2期临床试验(NCT06800313)中,第一位转移性去雄抵抗性前列腺癌(mCRPC)患者接受了HLD-0915,一种雄激素受体(AR)靶向RIPTAC。(33)这项首次人体临床试验的目的是评估口服单药HLD-0915的安全性、耐受性、药代动力学(PK)、药效学(PD)和抗肿瘤活性。(33,34)该研究包括初始的1期临床试验和随后的2期临床试验。1期临床试验是一项剂量递增研究,以确定HLD-0915作为单药治疗的最大耐受剂量(MTD)和/或推荐剂量(s)。2期扩展队列旨在进一步评估HLD-0915的有效性和安全性。该1/2期临床研究计划招募多达80名mCRPC患者。HLD-0915是一种异双功能分子,由AR配体作为TP, EP配体和连接连接体组成。到目前为止,HLD-0915的EP和化学结构尚未公开。HLD-0915在包括耐药模型在内的临床前前列腺癌模型中口服有效,使肿瘤显著缩小,前列腺特异性抗原(PSA)降低,同时显示出良好的治疗指标。前列腺癌是全球第二大最常见的恶性肿瘤,也是男性癌症死亡的第五大原因,2022年约有150万新病例和39.7万人死亡。 (35)几乎所有前列腺癌患者都对AR信号抑制剂产生耐药性,这是由多种异质旁路耐药机制驱动的,包括AR基因突变和AR水平表达增加。在80%以上的mCRPC患者中,编码AR的AR基因或DNA的上游增强子区异常扩增。(36)前列腺癌患者的临床需求仍未得到满足,特别是那些晚期、耐药和最致命类型的前列腺癌患者。因此,迫切需要新的治疗方法来治疗这些具有挑战性的疾病。ar靶向的RIPTACs(如HLD-0915)具有独特的作用机制,正在成为一种有前景的mCRPC的替代精确癌症治疗方法。为了开发治疗对AR拮抗剂(如enzalutamide)耐药的前列腺癌的新疗法,Halda therapeutics的研究人员设计并合成了一系列靶向AR的异双功能分子。令人兴奋的是,两种化合物H001和H003被证明是治疗前列腺癌的有希望的候选者。(33) H001和H003是真正的riptac,通过化学诱导的接近机制起作用。H001和H003与TP (AR)和EP形成稳定的三元配合物,这是由AR:RIPTAC:EP三元配合物的x射线共晶结构揭示的,但PDB代码尚未公开。AR:RIPTAC:EP三元配合物的x射线晶体结构的确定使AR靶向RIPTAC的基于结构的药物设计(SBDD)成为可能。H001和H003在VcaP细胞中与AR和EP形成胞内三元配合物,EC50值分别为24 nM和9 nM。然而,AR配体和EP配体都不能单独诱导细胞内三元复合物的形成,这表明riptac内的AR配体和EP配体都参与了三元复合物的形成。此外,AR靶向riptac在AR结合和TP结合中表现出积极的协同作用,有助于形成更稳定的三元配合物。H001和H003表现出较强的抗增殖活性(H001: IC50 <;10海里;H003: IC50;与表达低水平FL-AR的亲代22RV1细胞相比,H001和H003显著诱导过表达FL-AR的22RV1细胞凋亡(H001: EC50 = 113 nM;H003: EC50 = 48.5 nM),通过Caspase 3/7 GIo测定。过表达FL-AR的22RV1/AR细胞来源于亲代AR- v7 +细胞,这是一种enzalutamide耐药细胞系。H001在多西环素诱导的表达临床相关AR突变(ARMut)的Trex293细胞中诱导AR:RIPTAC:EP三元复合物的形成。多西环素诱导的ARMut导致702位赖氨酸残基被组氨酸(L702H)取代,875位组氨酸残基被酪氨酸(H875Y)取代,878位苏氨酸残基被丙氨酸(T878A)取代。在去势小鼠AR扩增(ARamp)、V7+ VCaP前列腺肿瘤模型中,H001和H003 (30 mg/kg, p.o, QD)口服体内疗效优于Enzalutamide (AR信号抑制剂)。H001和H003在阉割小鼠VCaP肿瘤模型(恩杂鲁胺不敏感前列腺癌模型)中引起肿瘤消退。此外,H001和H003显著降低血浆PSA。此外,H001和H003在肿瘤组织中可与AR和EP形成三元配合物,抑制EP,转化为强大的体内药效。在阉割小鼠VCaP肿瘤模型中,预先给药占据AR配体结合域(AR- lbd)的Enzalutamide可竞争AR与H001的结合,导致三元复合物(AR:H001:EP)形成的显著衰减和EP抑制。这一发现表明H001的体内药效学(PD)调节依赖于AR结合。H001在体外和体内VCaP模型中均诱导“BRCAness”。H001显著抑制乳腺癌基因1 (BRCA1)、乳腺癌基因2 (BRCA2)和相关同源重组修复(HRR)基因,如rad51相关蛋白1 (RAD51AP1)、recq介导的基因组不稳定蛋白2 (RMI2)和rad54样蛋白(RAD54L)在VCaP细胞中的表达,并呈剂量依赖性。此外,H001 (30 mg/kg)可有效抑制去势VCaP肿瘤模型中这些HRR基因的表达。PARP抑制剂可导致BRCA1/2缺乏症前列腺癌细胞的合成致死。因此,H001与PARP抑制剂联合治疗前列腺癌可能发挥协同作用,显示出克服PARP抑制剂耐药的潜力。AR-RIPTACs和PARP抑制剂的联合治疗研究将被期待。与现有的治疗方式相比,riptac有几个独特的特点,如表1所示。 首先,RIPTACs独立于TP发挥功能,与双特异性抗体(bsAbs)、抗体-药物偶联物(adc)和CAR-T-mAb类似,但与小分子抑制剂和蛋白质降解剂不同。其次,RIPTACs对非靶向耐药机制引起的耐药癌细胞有效,这与bsab、adc和CAR-T-mAb类似,但与小分子抑制剂和蛋白质降解剂不同。第三,RIPTACs通过靶向肿瘤驱动的“不可药物”癌蛋白显示出治疗癌症的潜力,这类似于蛋白质降解剂、bsab、adc和CAR-T-mAb,但优于小分子抑制剂。第四,RIPTAC可诱导TP:RIPTAC:EP三元复合物的形成,诱导或增强TP与EP之间的PPIs,其作用类似于蛋白降解剂,如分子胶降解剂(MGDs),但与小分子抑制剂、bsab、adc、CAR-T-mAb不同。第五,与小分子抑制剂和蛋白质降解剂一样,RIPTACs可以作为口服剂量开发,具有低销售成本(COGS),这是bsab、adc和CAR-T-mAb无法获得的。此外,与小分子抑制剂和蛋白质降解剂一样,RIPTACs靶向细胞内TPs,这与bsab、adc和CAR-T-mAb依赖于细胞膜上的抗原不同。与现有的治疗方式相比,riptac具有几个优势。首先,RIPTACs选择性杀死癌细胞,同时保留正常细胞,提供高治疗指数。其次,riptac独立于TP功能而起作用,极大地扩展了TP的作用范围。为此,一些在肿瘤细胞中特异性表达的“不可药物”靶点可以作为tp用于开发riptac。第三,基于正合作结合机制,RIPTACs不需要与TP具有高结合亲和力,显示出克服TP突变引起的耐药的潜力。此外,riptac还具有克服非靶向耐药机制引起的耐药的潜力。然而,riptac的发展也面临着挑战。与PROTACs一样,riptac也具有相对较大的分子量,这超出了Lipinski的“五法则”(Ro5),限制了其整体的药物性质,如细胞渗透性、水溶性和代谢稳定性。通过系统和持续的结构优化,PROTACs的积极临床结果部分减轻了这种担忧。此外,HLD-0915的成功进入临床试验,激发了RIPTAC模式对药物开发的努力。由于其独特的分子胶机制,riptac对TP和EP的配体选择以及连接体的类型和长度的要求比PROTACs更严格。因此,riptac的开发成功率低于PROTACs。设计和合成的作为潜在riptac的二价异双功能分子很少是真正的riptac,而大多数可能只是作为潜在的双重抑制剂而不诱导接近。此外,开发riptac的TPs数量在很大程度上是有限的。目前,只有AR、ER、FKBP和P53 Y220C蛋白被确定为适合riptac的TPs。(15,16,37,38)为了开发新的riptac,需要探索更多样化的TPs。(38)用于riptac的EPs主要包括细胞周期蛋白依赖性激酶(CDKs)、溴域和额外末端结构域(BET)蛋白和polo样激酶1 (PLK1)。(15,16)此外,RIPTACs目前主要用于治疗癌症,而这一策略也可能在未来扩展并适用于其他威胁疾病。总之,RIPTAC技术是一种新的基于化学诱导的接近性治疗策略,为创新药物的发现和开发开辟了新的途径。riptac具有独特的选择性和广泛适用的癌细胞杀伤机制,有可能减轻副作用,包括靶外组织毒性和克服耐药性。作为一项新兴技术,RIPTAC的开发仍处于早期阶段,但受首个RIPTAC HLD-0915成功推进到治疗mCRPC (NCT06800313)的1/2期临床试验这一里程碑的启发。(34)虽然我们期待这些研究的积极临床结果,但RIPTACs作为一种新的模式可能为开发精准药物,特别是癌症治疗提供无与伦比的机会。这项工作得到了美国国立卫生研究院R01CA226001和R01CA231150拨款的部分支持,乳腺癌研究计划(BCRP)突破奖W81XWH-17-1-0071和W81XWH-17-1-0072来自国防部(DoD), John D. Stobo, M.D.杰出主席捐赠基金和Edith &;罗伯特·津恩,药物发现基金主席。本文引用了38个其他出版物。这篇文章尚未被其他出版物引用。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Journal of Medicinal Chemistry
医学-医药化学
CiteScore
4.00
自引率
11.00%
发文量
804
审稿时长
1.9 months
期刊介绍:
The Journal of Medicinal Chemistry is a prestigious biweekly peer-reviewed publication that focuses on the multifaceted field of medicinal chemistry. Since its inception in 1959 as the Journal of Medicinal and Pharmaceutical Chemistry, it has evolved to become a cornerstone in the dissemination of research findings related to the design, synthesis, and development of therapeutic agents.
The Journal of Medicinal Chemistry is recognized for its significant impact in the scientific community, as evidenced by its 2022 impact factor of 7.3. This metric reflects the journal's influence and the importance of its content in shaping the future of drug discovery and development. The journal serves as a vital resource for chemists, pharmacologists, and other researchers interested in the molecular mechanisms of drug action and the optimization of therapeutic compounds.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


