Jean-Daniel Masson, Valentina Taglietti, François Ruby, Hiroya Ono, Nadir Mouri, Alan Jorge, Laurent Guillaud, Laurent Tiret, Frederic Relaix
{"title":"Extensive striated muscle damage in a rat model of Duchenne muscular dystrophy with Dmd exons 10-17 duplication.","authors":"Jean-Daniel Masson, Valentina Taglietti, François Ruby, Hiroya Ono, Nadir Mouri, Alan Jorge, Laurent Guillaud, Laurent Tiret, Frederic Relaix","doi":"10.1186/s13395-025-00386-2","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Duchenne muscular dystrophy (DMD) mainly affects young boys with out-of-frame mutations in the DMD gene, leading to dystrophin deficiency. This loss disrupts the assembly of the sarcolemmal dystrophin-associated glycoprotein complex, resulting in membrane fragility and damage during muscle contraction-relaxation cycles. Consequently, patients experience progressive muscle weakness, loss of ambulation and cardiorespiratory failure. Gene therapy represents one of the most promising therapeutic approaches, requiring rigorous preclinical validation of candidate strategies. While several preclinical models of dystrophin deficiency mimic point mutations or exon deletions, no existing rat model accurately replicates DMD gene duplications, which account for approximately 10% of DMD cases.</p><p><strong>Methods: </strong>Using CRISPR/Cas9 genome editing, we generated a ~ 125 kbp duplication encompassing exons 10-17 of the Dmd gene in Sprague Dawley rats. To characterise disease progression in these rats, we assessed biochemical, histological and functional biomarkers at 6 and 10 months of age, comparing them to their healthy littermates.</p><p><strong>Results: </strong>We established the R-DMDdup10-17 line. The microstructure of limb, diaphragm and cardiac muscles of R-DMDdup10-17 (DMD) rats exhibited dystrophic changes at 6 and 10 months, including loss of myofibres and fibrosis. These alterations led to a significant body mass reduction, muscle weakness (including diaphragm deficiency) and cardiac electrical defects. Premature lethality was observed between 10 and 13 months.</p><p><strong>Conclusion: </strong>Duplication of the Dmd genomic region encompassing exons 10 to 17 in rats results in dystrophin deficiency, severe striated muscle dystrophy, and premature death. The R-DMDdup10-17 line represents the first reported genetic model of a severe and early lethal duplication variant in the Dmd gene. It provides a critical tool for assessing targeted gene therapies aimed to correct such mutations.</p>","PeriodicalId":21747,"journal":{"name":"Skeletal Muscle","volume":"15 1","pages":"16"},"PeriodicalIF":4.4000,"publicationDate":"2025-06-09","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12147255/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Skeletal Muscle","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s13395-025-00386-2","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CELL BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Duchenne muscular dystrophy (DMD) mainly affects young boys with out-of-frame mutations in the DMD gene, leading to dystrophin deficiency. This loss disrupts the assembly of the sarcolemmal dystrophin-associated glycoprotein complex, resulting in membrane fragility and damage during muscle contraction-relaxation cycles. Consequently, patients experience progressive muscle weakness, loss of ambulation and cardiorespiratory failure. Gene therapy represents one of the most promising therapeutic approaches, requiring rigorous preclinical validation of candidate strategies. While several preclinical models of dystrophin deficiency mimic point mutations or exon deletions, no existing rat model accurately replicates DMD gene duplications, which account for approximately 10% of DMD cases.
Methods: Using CRISPR/Cas9 genome editing, we generated a ~ 125 kbp duplication encompassing exons 10-17 of the Dmd gene in Sprague Dawley rats. To characterise disease progression in these rats, we assessed biochemical, histological and functional biomarkers at 6 and 10 months of age, comparing them to their healthy littermates.
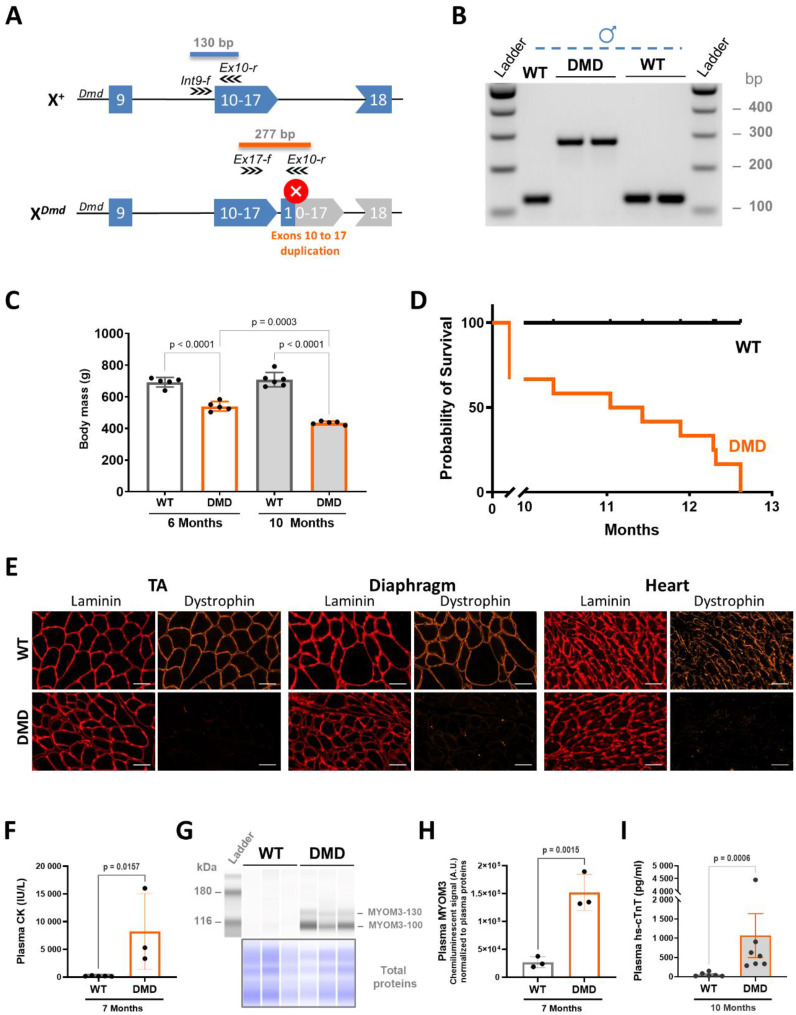
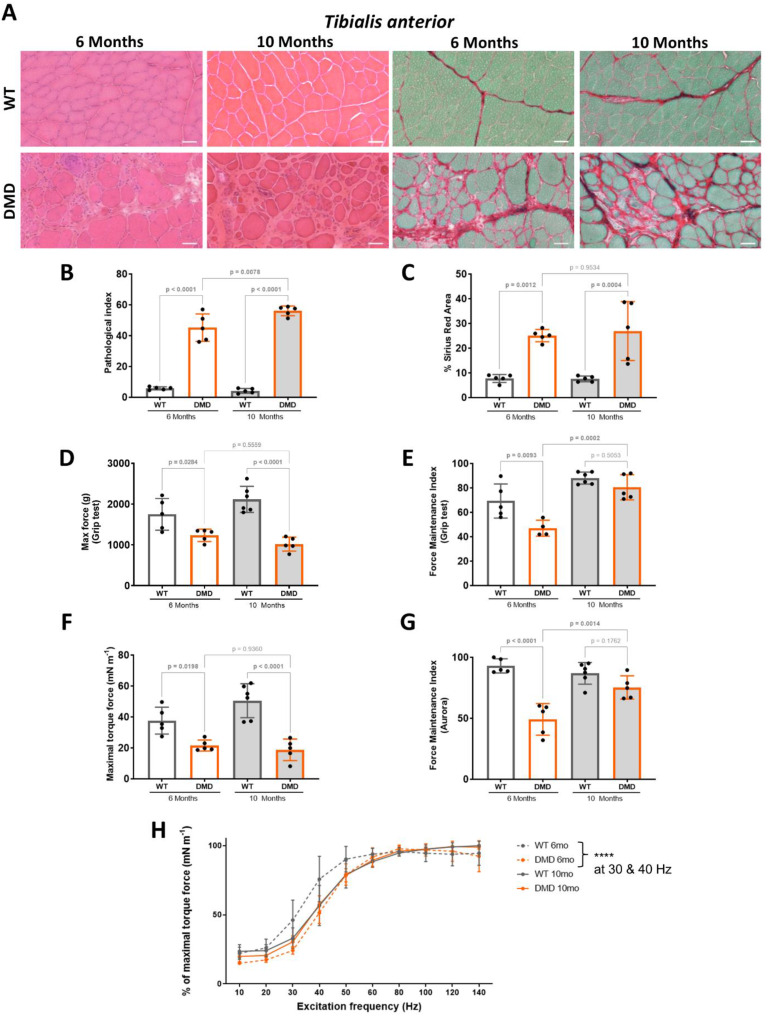
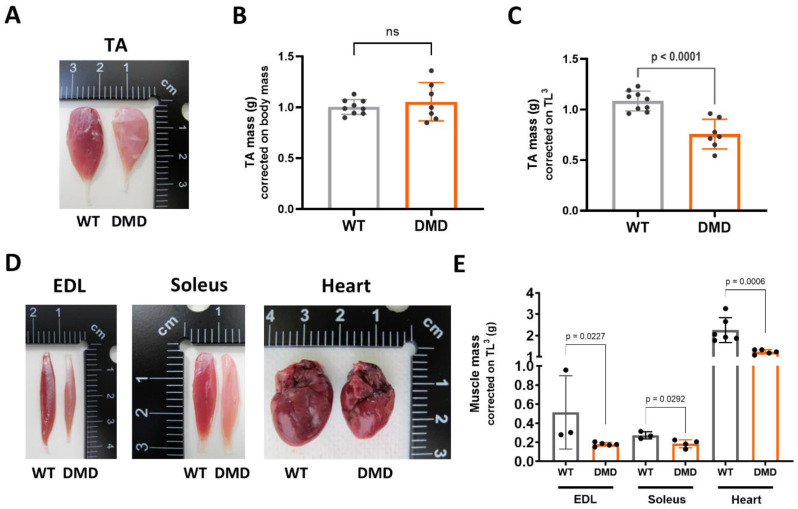
Results: We established the R-DMDdup10-17 line. The microstructure of limb, diaphragm and cardiac muscles of R-DMDdup10-17 (DMD) rats exhibited dystrophic changes at 6 and 10 months, including loss of myofibres and fibrosis. These alterations led to a significant body mass reduction, muscle weakness (including diaphragm deficiency) and cardiac electrical defects. Premature lethality was observed between 10 and 13 months.
Conclusion: Duplication of the Dmd genomic region encompassing exons 10 to 17 in rats results in dystrophin deficiency, severe striated muscle dystrophy, and premature death. The R-DMDdup10-17 line represents the first reported genetic model of a severe and early lethal duplication variant in the Dmd gene. It provides a critical tool for assessing targeted gene therapies aimed to correct such mutations.
期刊介绍:
The only open access journal in its field, Skeletal Muscle publishes novel, cutting-edge research and technological advancements that investigate the molecular mechanisms underlying the biology of skeletal muscle. Reflecting the breadth of research in this area, the journal welcomes manuscripts about the development, metabolism, the regulation of mass and function, aging, degeneration, dystrophy and regeneration of skeletal muscle, with an emphasis on understanding adult skeletal muscle, its maintenance, and its interactions with non-muscle cell types and regulatory modulators.
Main areas of interest include:
-differentiation of skeletal muscle-
atrophy and hypertrophy of skeletal muscle-
aging of skeletal muscle-
regeneration and degeneration of skeletal muscle-
biology of satellite and satellite-like cells-
dystrophic degeneration of skeletal muscle-
energy and glucose homeostasis in skeletal muscle-
non-dystrophic genetic diseases of skeletal muscle, such as Spinal Muscular Atrophy and myopathies-
maintenance of neuromuscular junctions-
roles of ryanodine receptors and calcium signaling in skeletal muscle-
roles of nuclear receptors in skeletal muscle-
roles of GPCRs and GPCR signaling in skeletal muscle-
other relevant aspects of skeletal muscle biology.
In addition, articles on translational clinical studies that address molecular and cellular mechanisms of skeletal muscle will be published. Case reports are also encouraged for submission.
Skeletal Muscle reflects the breadth of research on skeletal muscle and bridges gaps between diverse areas of science for example cardiac cell biology and neurobiology, which share common features with respect to cell differentiation, excitatory membranes, cell-cell communication, and maintenance. Suitable articles are model and mechanism-driven, and apply statistical principles where appropriate; purely descriptive studies are of lesser interest.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


