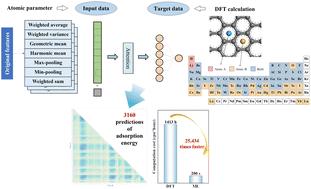
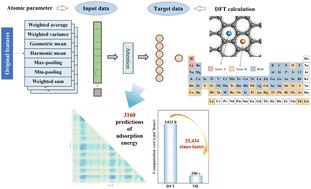
Integration of a self-attentive neural network and density functional theory for accelerated screening of graphene-based stabilized binary adsorption systems†
{"title":"Integration of a self-attentive neural network and density functional theory for accelerated screening of graphene-based stabilized binary adsorption systems†","authors":"Jiaying Chen, Yong Liu, Longlong Dong, Longfei Guo, Jingteng Xue, Zongfan Wei and Jingchuan Zhu","doi":"10.1039/D5TA01512J","DOIUrl":null,"url":null,"abstract":"<p >It is crucial to study adsorbed atomic systems in graphene as exogenous atoms or molecules can precisely modulate the properties of graphene and significantly extend its wide-range applications. However, graphene's exceptional sensitivity to environmental conditions complicates the precise control of the spatial distributions and binding modes of adsorbates at the atomic scale. Moreover, the stability and reversibility of these systems often rely on rigorous preparation processes and characterization methods. Currently, no effective method exists for systematically identifying and screening graphene adsorption systems with optimal performance. To tackle these challenges, herein, we integrate first principle calculations with machine learning not only to accelerate material screening and structural design but also to elucidate the key influencing factors of graphene adsorption systems. Our studies reveal that combining first principles with a self-attentive neural network can successfully predict the stability of adsorption systems of the previously unseen graphene samples. Furthermore, the trained self-attentive neural network model predicts the adsorption energy trends of diatom systems involving transition metals and rare earth metals, while clarifying the effects of electronic structures, d orbital occupancy, and ionic radius of the elements on their adsorption behavior. This research is expected to advance the design and optimization of new generation graphene-based electronic devices, sensors, and catalytic systems, providing more efficient and precise pathways for future material designs.</p>","PeriodicalId":82,"journal":{"name":"Journal of Materials Chemistry A","volume":" 26","pages":" 21021-21034"},"PeriodicalIF":9.5000,"publicationDate":"2025-06-09","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Materials Chemistry A","FirstCategoryId":"88","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2025/ta/d5ta01512j","RegionNum":2,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
It is crucial to study adsorbed atomic systems in graphene as exogenous atoms or molecules can precisely modulate the properties of graphene and significantly extend its wide-range applications. However, graphene's exceptional sensitivity to environmental conditions complicates the precise control of the spatial distributions and binding modes of adsorbates at the atomic scale. Moreover, the stability and reversibility of these systems often rely on rigorous preparation processes and characterization methods. Currently, no effective method exists for systematically identifying and screening graphene adsorption systems with optimal performance. To tackle these challenges, herein, we integrate first principle calculations with machine learning not only to accelerate material screening and structural design but also to elucidate the key influencing factors of graphene adsorption systems. Our studies reveal that combining first principles with a self-attentive neural network can successfully predict the stability of adsorption systems of the previously unseen graphene samples. Furthermore, the trained self-attentive neural network model predicts the adsorption energy trends of diatom systems involving transition metals and rare earth metals, while clarifying the effects of electronic structures, d orbital occupancy, and ionic radius of the elements on their adsorption behavior. This research is expected to advance the design and optimization of new generation graphene-based electronic devices, sensors, and catalytic systems, providing more efficient and precise pathways for future material designs.
期刊介绍:
The Journal of Materials Chemistry A, B & C covers a wide range of high-quality studies in the field of materials chemistry, with each section focusing on specific applications of the materials studied. Journal of Materials Chemistry A emphasizes applications in energy and sustainability, including topics such as artificial photosynthesis, batteries, and fuel cells. Journal of Materials Chemistry B focuses on applications in biology and medicine, while Journal of Materials Chemistry C covers applications in optical, magnetic, and electronic devices. Example topic areas within the scope of Journal of Materials Chemistry A include catalysis, green/sustainable materials, sensors, and water treatment, among others.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


