Unravelling the genomes of multidrug-resistant Klebsiella quasipneumoniae: A study of clinical and environmental isolates
IF 2.6
4区 医学
Q3 INFECTIOUS DISEASES
引用次数: 0
Abstract
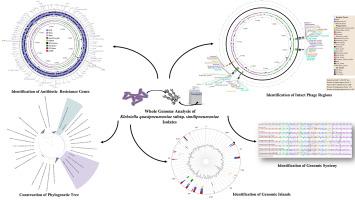
Klebsiella quasipneumoniae is an opportunistic pathogen that predominantly resides in the human gut posing a significant risk of severe infections in individuals with compromised immune systems. In this study, whole genome sequencing of two multi-drug-resistant clinical and freshwater K. quasipneumoniae subsp. similipneumoniae isolates was performed and compared using Illumina sequencing technology. The genome size of the clinical isolate and freshwater isolate was 5.23 Mbp and 5.22 Mbp, respectively, with a Guanine-cytosine (GC) content of 57.77 % and 57.21 %. Genomic analyses identified 29 genes associated with antimicrobial resistance, mainly related to efflux pumps. CRISPR sequences were also predicted, of which 4 were identified in the freshwater isolate and 1 in the clinical isolate. Genomic islets (GIts) and genomic islands (GIs) were also delineated using IslandViewer4. The freshwater isolate contained 13 GIs and 16 GIts, while the clinical isolate contained 14 GIs and 19 GIts, harbouring important virulence and antimicrobial resistance genes (such as acr, bdcA, fim and norB). PHASTEST analysis revealed six intact phage regions in the freshwater isolate and five in the clinical isolate. Finally, a Maximum Likelihood tree was constructed based on the amino acid sequences of 440 orthologous genes from the 98 K. quasipneumoniae genomes, showing that the isolates were positioned within distinct internal clades.
多重耐药准肺炎克雷伯菌基因组的揭示:临床和环境分离株的研究
准肺炎克雷伯菌是一种主要存在于人体肠道的机会性病原体,对免疫系统受损的个体造成严重感染的重大风险。本研究对两种多重耐药临床和淡水拟肺炎克雷伯菌亚种进行全基因组测序。采用Illumina测序技术对分离的类似肺炎菌进行分析和比较。临床分离株和淡水分离株基因组大小分别为5.23 Mbp和5.22 Mbp,鸟嘌呤-胞嘧啶(GC)含量分别为57.77%和57.21%。基因组分析确定了29个与抗菌素耐药性相关的基因,主要与外排泵有关。预测了4个淡水分离株和1个临床分离株的CRISPR序列。基因组岛和基因组岛也用islandviewer进行了划分。淡水分离株含有13个GIs和16个git,临床分离株含有14个GIs和19个git,含有重要的毒力和耐药基因(如acr、bdcA、film和norB)。phasest分析显示淡水分离株中有6个完整的噬菌体区域,临床分离株中有5个完整的噬菌体区域。最后,基于98份拟肺炎克雷伯菌基因组440个同源基因的氨基酸序列构建了极大似然树,结果表明分离株位于不同的内部进化支中。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Infection Genetics and Evolution
医学-传染病学
CiteScore
8.40
自引率
0.00%
发文量
215
审稿时长
82 days
期刊介绍:
(aka Journal of Molecular Epidemiology and Evolutionary Genetics of Infectious Diseases -- MEEGID)
Infectious diseases constitute one of the main challenges to medical science in the coming century. The impressive development of molecular megatechnologies and of bioinformatics have greatly increased our knowledge of the evolution, transmission and pathogenicity of infectious diseases. Research has shown that host susceptibility to many infectious diseases has a genetic basis. Furthermore, much is now known on the molecular epidemiology, evolution and virulence of pathogenic agents, as well as their resistance to drugs, vaccines, and antibiotics. Equally, research on the genetics of disease vectors has greatly improved our understanding of their systematics, has increased our capacity to identify target populations for control or intervention, and has provided detailed information on the mechanisms of insecticide resistance.
However, the genetics and evolutionary biology of hosts, pathogens and vectors have tended to develop as three separate fields of research. This artificial compartmentalisation is of concern due to our growing appreciation of the strong co-evolutionary interactions among hosts, pathogens and vectors.
Infection, Genetics and Evolution and its companion congress [MEEGID](http://www.meegidconference.com/) (for Molecular Epidemiology and Evolutionary Genetics of Infectious Diseases) are the main forum acting for the cross-fertilization between evolutionary science and biomedical research on infectious diseases.
Infection, Genetics and Evolution is the only journal that welcomes articles dealing with the genetics and evolutionary biology of hosts, pathogens and vectors, and coevolution processes among them in relation to infection and disease manifestation. All infectious models enter the scope of the journal, including pathogens of humans, animals and plants, either parasites, fungi, bacteria, viruses or prions. The journal welcomes articles dealing with genetics, population genetics, genomics, postgenomics, gene expression, evolutionary biology, population dynamics, mathematical modeling and bioinformatics. We also provide many author benefits, such as free PDFs, a liberal copyright policy, special discounts on Elsevier publications and much more. Please click here for more information on our author services .
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


