{"title":"Dilated Cardiomyopathy May Be Associated With a Novel Mitochondrial tRNASer(AGY) Mutation","authors":"Yu Ding, Xuejiao Yu, Jian Xu, Shunrong Zhang, Jianhang Leng","doi":"10.1155/humu/7888334","DOIUrl":null,"url":null,"abstract":"<p>Dilated cardiomyopathy (DCM) is a serious public health problem that increases the risk of developing heart failure. Most recently, increasing evidence has shown that mitochondrial dysfunction caused by mitochondrial tRNA (mt-tRNA) mutations plays a putative role in the pathogenesis of this disease, despite its pathophysiology remaining poorly understood. In this study, a novel 12265A>G mutation in mt-tRNA<sup>Ser(AGY)</sup> was identified from a Chinese pedigree with maternally inherited DCM, together with a known mt-tRNA<sup>Cys</sup> 5821G>A mutation. Interestingly, the novel m.12265A>G mutation changed the well-conserved adenosine at Position 73 (A73) to guanine (G73) at the 3 <sup>′</sup>-end of the mt-tRNA<sup>Ser(AGY)</sup> acceptor arm, while the G-to-A transition at 5821 occurred at the acceptor arm of mt-tRNA<sup>Cys</sup>, disrupting conserved base pairing (G6-C67). Transmitochondrial cybrid-based study demonstrated that cell lines with m.12265A>G and m.5821G>A mutations showed impaired mitochondrial functions, including significant reductions in mitochondrial ATP, membrane potential, NAD<sup>+</sup>/NADH ratio, mitochondrial DNA (mtDNA) content, mitochondrial transcription factor A (TFAM) mRNA expression levels, and respiratory chain enzyme Complex I and III activities, whereas the levels of reactive oxygen species (ROS), calcium ions (Ca<sup>2+</sup>), and lactate were enhanced in mutant cells compared to controls (<i>p</i> < 0.05). Thus, the m.12265A>G and m.5821G>A mutations may affect mt-tRNA metabolism and impair mitochondrial function, which is involved in DCM. Taken together, our study broadens the genotypic interpretation of mt-tRNA mutations linked to disease.</p>","PeriodicalId":13061,"journal":{"name":"Human Mutation","volume":"2025 1","pages":""},"PeriodicalIF":3.7000,"publicationDate":"2025-06-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1155/humu/7888334","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Human Mutation","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1155/humu/7888334","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
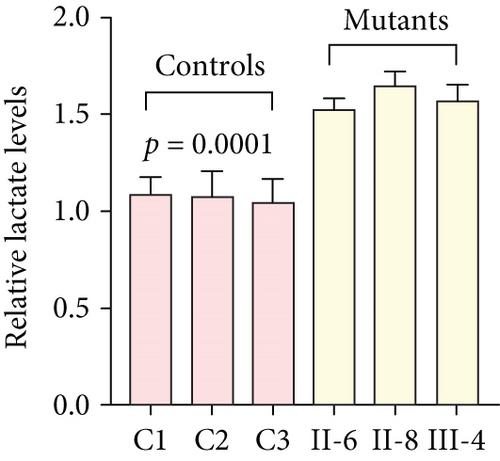
Dilated cardiomyopathy (DCM) is a serious public health problem that increases the risk of developing heart failure. Most recently, increasing evidence has shown that mitochondrial dysfunction caused by mitochondrial tRNA (mt-tRNA) mutations plays a putative role in the pathogenesis of this disease, despite its pathophysiology remaining poorly understood. In this study, a novel 12265A>G mutation in mt-tRNASer(AGY) was identified from a Chinese pedigree with maternally inherited DCM, together with a known mt-tRNACys 5821G>A mutation. Interestingly, the novel m.12265A>G mutation changed the well-conserved adenosine at Position 73 (A73) to guanine (G73) at the 3 ′-end of the mt-tRNASer(AGY) acceptor arm, while the G-to-A transition at 5821 occurred at the acceptor arm of mt-tRNACys, disrupting conserved base pairing (G6-C67). Transmitochondrial cybrid-based study demonstrated that cell lines with m.12265A>G and m.5821G>A mutations showed impaired mitochondrial functions, including significant reductions in mitochondrial ATP, membrane potential, NAD+/NADH ratio, mitochondrial DNA (mtDNA) content, mitochondrial transcription factor A (TFAM) mRNA expression levels, and respiratory chain enzyme Complex I and III activities, whereas the levels of reactive oxygen species (ROS), calcium ions (Ca2+), and lactate were enhanced in mutant cells compared to controls (p < 0.05). Thus, the m.12265A>G and m.5821G>A mutations may affect mt-tRNA metabolism and impair mitochondrial function, which is involved in DCM. Taken together, our study broadens the genotypic interpretation of mt-tRNA mutations linked to disease.
期刊介绍:
Human Mutation is a peer-reviewed journal that offers publication of original Research Articles, Methods, Mutation Updates, Reviews, Database Articles, Rapid Communications, and Letters on broad aspects of mutation research in humans. Reports of novel DNA variations and their phenotypic consequences, reports of SNPs demonstrated as valuable for genomic analysis, descriptions of new molecular detection methods, and novel approaches to clinical diagnosis are welcomed. Novel reports of gene organization at the genomic level, reported in the context of mutation investigation, may be considered. The journal provides a unique forum for the exchange of ideas, methods, and applications of interest to molecular, human, and medical geneticists in academic, industrial, and clinical research settings worldwide.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


