Alma D Méndez-Álvarez, Karla P Meneses-León, Dira B Monsalvo-Soler, Alondra M Morales-Segundo, Laura Gómez-Virgilio, Gustavo López-Toledo
{"title":"[Landau-Kleffner Syndrome: Current Etiopathogenesis and Management].","authors":"Alma D Méndez-Álvarez, Karla P Meneses-León, Dira B Monsalvo-Soler, Alondra M Morales-Segundo, Laura Gómez-Virgilio, Gustavo López-Toledo","doi":"10.31083/RN42643","DOIUrl":null,"url":null,"abstract":"<p><p>Landau-Kleffner syndrome is a developmental epileptic encephalopathy that manifests mainly in pediatric patients, characterized by verbal auditory agnosia and focal, bilateral, and focal and diffuse epileptic activity, visualized through electroencephalographic recordings performed during sleep. It is a rare syndrome with a variable, multifactorial presentation and unknown etiology, although it has a genetic component in some cases. It is often associated with variants of the glutamate ionotropic receptor N-methyl-D-aspartate (NMDA) type subunit 2A (GRIN2A) gene, which encodes an NMDA receptor subunit of the same name that is involved in various neurophysiological processes. Modifications to this receptor could be associated with the clinical manifestations observed in patients. This review proposes a pathophysiological mechanism related to one of the clinical presentations of this disease, using information published in recent years, and contributes to the understanding of its pathology and the improvement of its management. This syndrome is a rare and complex disease; both its diagnosis and treatment are challenging, limiting patients' therapeutic options and compromising their quality of life.</p>","PeriodicalId":21281,"journal":{"name":"Revista de neurologia","volume":"80 4","pages":"42643"},"PeriodicalIF":0.8000,"publicationDate":"2025-05-27","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12135646/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Revista de neurologia","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.31083/RN42643","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
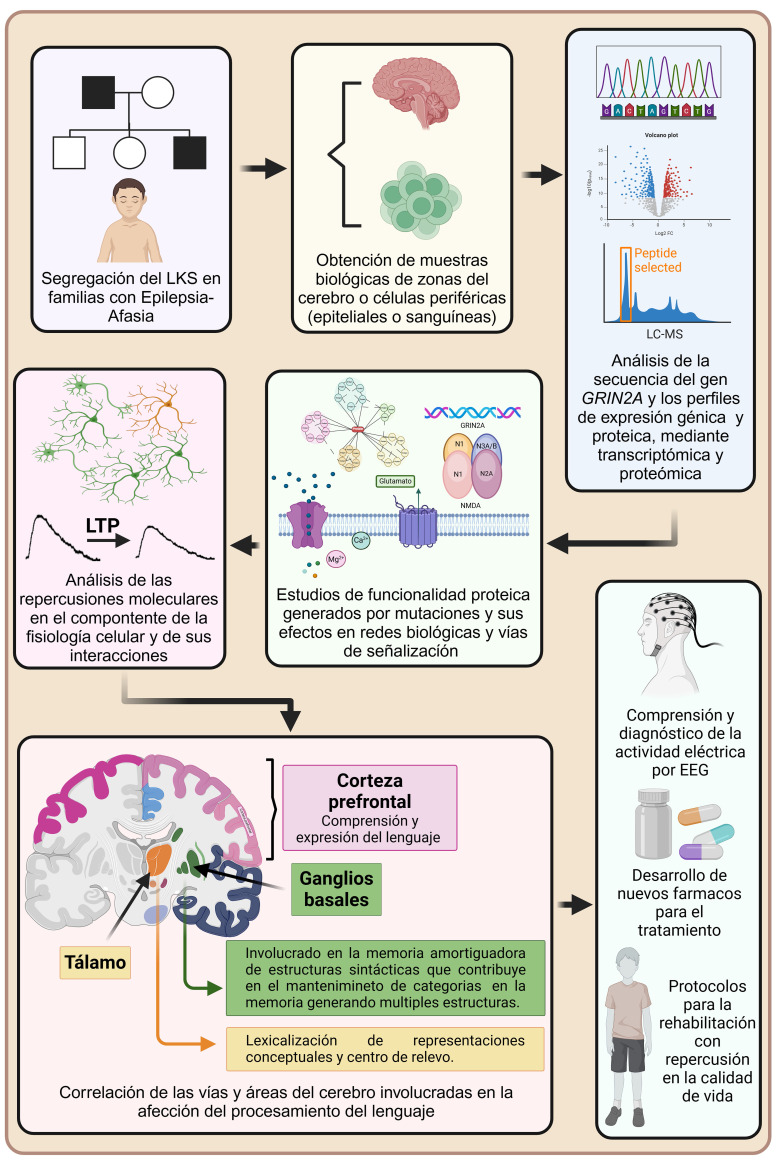
Landau-Kleffner syndrome is a developmental epileptic encephalopathy that manifests mainly in pediatric patients, characterized by verbal auditory agnosia and focal, bilateral, and focal and diffuse epileptic activity, visualized through electroencephalographic recordings performed during sleep. It is a rare syndrome with a variable, multifactorial presentation and unknown etiology, although it has a genetic component in some cases. It is often associated with variants of the glutamate ionotropic receptor N-methyl-D-aspartate (NMDA) type subunit 2A (GRIN2A) gene, which encodes an NMDA receptor subunit of the same name that is involved in various neurophysiological processes. Modifications to this receptor could be associated with the clinical manifestations observed in patients. This review proposes a pathophysiological mechanism related to one of the clinical presentations of this disease, using information published in recent years, and contributes to the understanding of its pathology and the improvement of its management. This syndrome is a rare and complex disease; both its diagnosis and treatment are challenging, limiting patients' therapeutic options and compromising their quality of life.
Landau-Kleffner综合征是一种发展性癫痫性脑病,主要表现在儿科患者,其特征是言语听觉失认和局灶性、双侧性、局灶性和弥漫性癫痫活动,通过睡眠时的脑电图记录可见。这是一种罕见的综合征,具有可变的、多因素的表现和未知的病因,尽管在某些情况下它有遗传成分。它通常与谷氨酸嗜离子受体n -甲基- d -天冬氨酸(NMDA)型亚基2A (GRIN2A)基因的变异有关,该基因编码一个同名的NMDA受体亚基,参与各种神经生理过程。这种受体的修饰可能与患者观察到的临床表现有关。本文结合近年来发表的文献资料,对该疾病的一种临床表现提出了相关的病理生理机制,有助于对其病理的认识和治疗的改进。这种综合征是一种罕见而复杂的疾病;其诊断和治疗都具有挑战性,限制了患者的治疗选择并损害了他们的生活质量。

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


