Desiree Henares, Meritxell Cubero, Irene Martinez-de-Albeniz, Alba Arranz, Muntsa Rocafort, Pedro Brotons, Amaresh Perez-Argüello, Maria Jose Troyano, Amadeu Gene, Aleix Lluansi, Marti Iriondo-Sanz, Iolanda Jordan, Claudia Fortuny, Mireia Urrea, C Muñoz-Almagro
{"title":"Rapid identification of a Serratia marcescens outbreak in a neonatal intensive care unit by third-generation long-read nanopore sequencing.","authors":"Desiree Henares, Meritxell Cubero, Irene Martinez-de-Albeniz, Alba Arranz, Muntsa Rocafort, Pedro Brotons, Amaresh Perez-Argüello, Maria Jose Troyano, Amadeu Gene, Aleix Lluansi, Marti Iriondo-Sanz, Iolanda Jordan, Claudia Fortuny, Mireia Urrea, C Muñoz-Almagro","doi":"10.1186/s13756-025-01582-x","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Serratia marcescens is a frequent cause of outbreaks in high-risk hospital settings such as neonatal intensive care units (NICU). This study investigated a potential S. marcescens outbreak in the NICU of a reference children's hospital using Whole Genome Sequencing (WGS). Additionally, it assessed the performance of third-generation sequencing for the rapid and accurate identification and characterization of the outbreak's clonal strain.</p><p><strong>Methods: </strong>A prospective study was conducted from September 8th to November 12th 2021, following a sharp increase in invasive S. marcescens infections in the NICU of University Children's Hospital Sant Joan de Déu (Barcelona, Spain). This study included all patients admitted to NICU and other hospital wards from whom S. marcescens was isolated in any sample type. Nanopore sequencing was performed on S. marcescens isolates. Genomic characterization included phylogenetic analyses and detection of antimicrobial resistance genes.</p><p><strong>Results: </strong>Twenty-nine patients (16 NICU and 13 non-NICU patients) infected/colonized by S. marcescens were detected during the study period, accounting for a total of 61 isolates. The genomic characterization was performed on 24 isolates from 14 NICU-patients and 10 isolates from eight non-NICU patients. Phylogenetic analyses evidenced three clusters of closely related strains; cluster I (n = 22), II (n = 2) and III (n = 5). The remaining isolates (n = 5) did not cluster. Cluster I contained most isolates from NICU patients (20/24), and most isolates from NICU-patients with confirmed invasive disease (7/8). Cluster II contained two isolates from two NICU-patients, one presenting with invasive disease. The resistance gene blaSRT was found in 97% of S. marcescens isolates (33/34). All isolates exhibited the amikacin-tobramycin aac(6') resistance gene and three multi-drug efflux pumps genes; sdeY, sdeB and smfY. The tetracycline tet(41) resistance gene was found in non-clustered isolates (4/34). The first results were available less than one month after the outbreak's alarm, and complete genomic study after two months.</p><p><strong>Conclusion: </strong>Two clonal strains were co-circulating in the NICU setting, with one being the major strain responsible for the outbreak. Rapid molecular characterization with nanopore sequencing confirmed the outbreak. It revealed the phylogenetic relationships among isolates and their antimicrobial potential. This approach enabled effective contextualization of the outbreak and allowed for monitoring its progression.</p>","PeriodicalId":7950,"journal":{"name":"Antimicrobial Resistance and Infection Control","volume":"14 1","pages":"63"},"PeriodicalIF":4.4000,"publicationDate":"2025-06-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12135216/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Antimicrobial Resistance and Infection Control","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s13756-025-01582-x","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"INFECTIOUS DISEASES","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Serratia marcescens is a frequent cause of outbreaks in high-risk hospital settings such as neonatal intensive care units (NICU). This study investigated a potential S. marcescens outbreak in the NICU of a reference children's hospital using Whole Genome Sequencing (WGS). Additionally, it assessed the performance of third-generation sequencing for the rapid and accurate identification and characterization of the outbreak's clonal strain.
Methods: A prospective study was conducted from September 8th to November 12th 2021, following a sharp increase in invasive S. marcescens infections in the NICU of University Children's Hospital Sant Joan de Déu (Barcelona, Spain). This study included all patients admitted to NICU and other hospital wards from whom S. marcescens was isolated in any sample type. Nanopore sequencing was performed on S. marcescens isolates. Genomic characterization included phylogenetic analyses and detection of antimicrobial resistance genes.
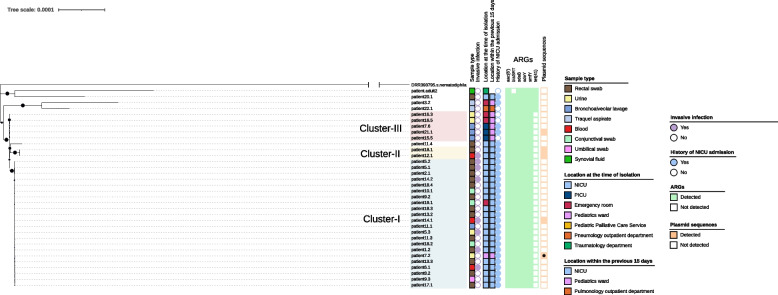
Results: Twenty-nine patients (16 NICU and 13 non-NICU patients) infected/colonized by S. marcescens were detected during the study period, accounting for a total of 61 isolates. The genomic characterization was performed on 24 isolates from 14 NICU-patients and 10 isolates from eight non-NICU patients. Phylogenetic analyses evidenced three clusters of closely related strains; cluster I (n = 22), II (n = 2) and III (n = 5). The remaining isolates (n = 5) did not cluster. Cluster I contained most isolates from NICU patients (20/24), and most isolates from NICU-patients with confirmed invasive disease (7/8). Cluster II contained two isolates from two NICU-patients, one presenting with invasive disease. The resistance gene blaSRT was found in 97% of S. marcescens isolates (33/34). All isolates exhibited the amikacin-tobramycin aac(6') resistance gene and three multi-drug efflux pumps genes; sdeY, sdeB and smfY. The tetracycline tet(41) resistance gene was found in non-clustered isolates (4/34). The first results were available less than one month after the outbreak's alarm, and complete genomic study after two months.
Conclusion: Two clonal strains were co-circulating in the NICU setting, with one being the major strain responsible for the outbreak. Rapid molecular characterization with nanopore sequencing confirmed the outbreak. It revealed the phylogenetic relationships among isolates and their antimicrobial potential. This approach enabled effective contextualization of the outbreak and allowed for monitoring its progression.
期刊介绍:
Antimicrobial Resistance and Infection Control is a global forum for all those working on the prevention, diagnostic and treatment of health-care associated infections and antimicrobial resistance development in all health-care settings. The journal covers a broad spectrum of preeminent practices and best available data to the top interventional and translational research, and innovative developments in the field of infection control.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


