Muhammad Aamir, Fahad Faizullah, Malik W Z Khan, Touba Azeem, Muhammad Awais Khan
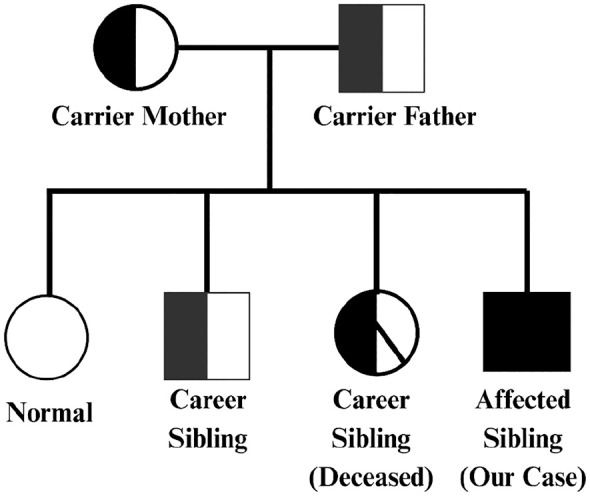
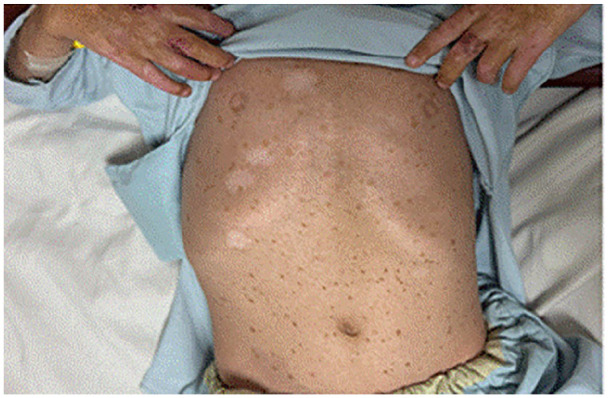
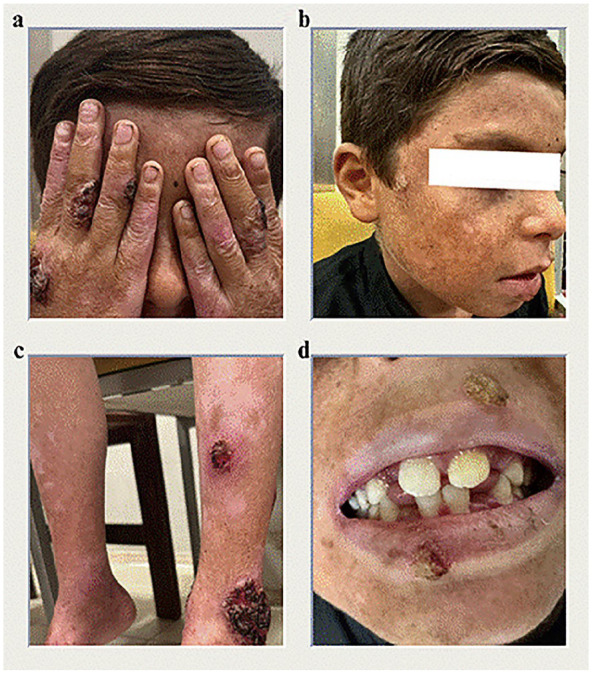
{"title":"Unique Dermatological and Systemic Manifestations in a Classic Pediatric Case of Kindler Syndrome: A Case Report and Literature Review.","authors":"Muhammad Aamir, Fahad Faizullah, Malik W Z Khan, Touba Azeem, Muhammad Awais Khan","doi":"10.1177/11795476251342637","DOIUrl":null,"url":null,"abstract":"<p><p>Kindler Syndrome (KS) is a rare, autosomal recessive genodermatosis caused by mutations in the FERMT1 gene, leading to skin fragility, blistering, photosensitivity, and progressive poikiloderma. We present a unique case of KS in a 6-year-old boy born to consanguineous parents, exhibiting uncommon dermatological, and systemic features. The patient developed multiple erythematous plaques, hemorrhagic crusting, and purulent discharge after birth, with a family history suggesting genetic predisposition. Uniquely, the patient presented with well-demarcated hyperpigmented macules on the abdomen, a feature rarely seen in KS, which adds to the phenotypic diversity of the condition. Additionally, the patient had extensive lanugo hair growth, nail dystrophy, and gingivitis, typical of KS, but without urinary or mucosal involvement, a departure from more classic presentations. The patient also presented with glucose intolerance, indicated by elevated glucose levels of 222 mg/dL, likely due to infection-induced metabolic dysregulation, which normalized after treatment. The differential diagnosis initially considered porphyria cutanea tarda (PCT) due to overlapping features like photosensitivity and skin fragility. However, laboratory findings, including normal liver function and the absence of specific PCT markers, effectively excluded PCT. Microbiological swabs from purulent discharge identified Staphylococcus aureus, which was sensitive to the prescribed antibiotics. Management focused on symptomatic relief with antibiotics, supportive care, and iron supplementation to address anemia caused by chronic skin erosions. The case highlights diagnostic challenges in resource-limited settings where genetic testing was unavailable. It underscores the need for heightened awareness of atypical KS manifestations, the importance of clinical evaluation and genetic counseling, and contributes to the expanding knowledge of KS, particularly in populations with consanguineous marriages.</p>","PeriodicalId":10357,"journal":{"name":"Clinical Medicine Insights. Case Reports","volume":"18 ","pages":"11795476251342637"},"PeriodicalIF":0.6000,"publicationDate":"2025-05-27","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12117226/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Clinical Medicine Insights. Case Reports","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1177/11795476251342637","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"Q3","JCRName":"MEDICINE, GENERAL & INTERNAL","Score":null,"Total":0}
引用次数: 0
Abstract
Kindler Syndrome (KS) is a rare, autosomal recessive genodermatosis caused by mutations in the FERMT1 gene, leading to skin fragility, blistering, photosensitivity, and progressive poikiloderma. We present a unique case of KS in a 6-year-old boy born to consanguineous parents, exhibiting uncommon dermatological, and systemic features. The patient developed multiple erythematous plaques, hemorrhagic crusting, and purulent discharge after birth, with a family history suggesting genetic predisposition. Uniquely, the patient presented with well-demarcated hyperpigmented macules on the abdomen, a feature rarely seen in KS, which adds to the phenotypic diversity of the condition. Additionally, the patient had extensive lanugo hair growth, nail dystrophy, and gingivitis, typical of KS, but without urinary or mucosal involvement, a departure from more classic presentations. The patient also presented with glucose intolerance, indicated by elevated glucose levels of 222 mg/dL, likely due to infection-induced metabolic dysregulation, which normalized after treatment. The differential diagnosis initially considered porphyria cutanea tarda (PCT) due to overlapping features like photosensitivity and skin fragility. However, laboratory findings, including normal liver function and the absence of specific PCT markers, effectively excluded PCT. Microbiological swabs from purulent discharge identified Staphylococcus aureus, which was sensitive to the prescribed antibiotics. Management focused on symptomatic relief with antibiotics, supportive care, and iron supplementation to address anemia caused by chronic skin erosions. The case highlights diagnostic challenges in resource-limited settings where genetic testing was unavailable. It underscores the need for heightened awareness of atypical KS manifestations, the importance of clinical evaluation and genetic counseling, and contributes to the expanding knowledge of KS, particularly in populations with consanguineous marriages.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


