Amber S E van Oirsouw, Michael A Hadders, Martijn Koetsier, Edith D J Peters, Nurit Assia Batzir, Tahsin Stefan Barakat, Diana Baralle, Adelyn Beil, Marie-Noëlle Bonnet-Dupeyron, Philip M Boone, Arjan Bouman, Deanna Alexis Carere, Benjamin Cogne, Leslie Dunnington, Laura S Farach, Casie A Genetti, Bertrand Isidor, Louis Januel, Aakash Joshi, Nayana Lahiri, Kristen N Lee, Idit Maya, Meriel McEntagart, Hope Northrup, Mathilde Pujalte, Kate Richardson, Susan Walker, Bobby P C Koeleman, Mariëlle Alders, Richard H van Jaarsveld, Renske Oegema
{"title":"KDM2B variants in the CxxC domain impair its DNA-binding ability and cause a distinct neurodevelopmental syndrome.","authors":"Amber S E van Oirsouw, Michael A Hadders, Martijn Koetsier, Edith D J Peters, Nurit Assia Batzir, Tahsin Stefan Barakat, Diana Baralle, Adelyn Beil, Marie-Noëlle Bonnet-Dupeyron, Philip M Boone, Arjan Bouman, Deanna Alexis Carere, Benjamin Cogne, Leslie Dunnington, Laura S Farach, Casie A Genetti, Bertrand Isidor, Louis Januel, Aakash Joshi, Nayana Lahiri, Kristen N Lee, Idit Maya, Meriel McEntagart, Hope Northrup, Mathilde Pujalte, Kate Richardson, Susan Walker, Bobby P C Koeleman, Mariëlle Alders, Richard H van Jaarsveld, Renske Oegema","doi":"10.1093/hmg/ddaf082","DOIUrl":null,"url":null,"abstract":"<p><p>Rare variants affecting the epigenetic regulator KDM2B cause a recently delineated neurodevelopmental disorder. Interestingly, we previously identified both a general KDM2B-associated episignature and a subsignature specific to variants in the DNA-binding CxxC domain. In light of the existence of a distinct subsignature, we set out to determine if KDM2B CxxC variants are associated with a unique phenotype and disease mechanism. We recruited individuals with heterozygous CxxC variants and assessed the variants' effect on protein expression and DNA-binding ability. We analyzed clinical data from 19 individuals, including ten previously undescribed individuals with seven novel CxxC variants. The core phenotype of the KDM2B-CxxC cohort is more extensive as compared to that of individuals with KDM2B haploinsufficiency. All individuals with CxxC variants presented with developmental delay, mainly in the speech and motor domain, in addition to variable intellectual disability and mild facial dysmorphism. Congenital heart defects were observed in up to 78% of individuals, with additional common findings including musculoskeletal, ophthalmological, and urogenital anomalies, as well as behavioral challenges and feeding difficulties. Functional assays revealed that while mutant KDM2B protein with CxxC variants can be expressed in vitro, its DNA-binding ability is significantly reduced compared to wildtype. This study shows that KDM2B CxxC variants cause a distinct neurodevelopmental syndrome, possibly through a molecular mechanism different from haploinsufficiency.</p>","PeriodicalId":13070,"journal":{"name":"Human molecular genetics","volume":" ","pages":"1353-1367"},"PeriodicalIF":3.2000,"publicationDate":"2025-08-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12361114/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Human molecular genetics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1093/hmg/ddaf082","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
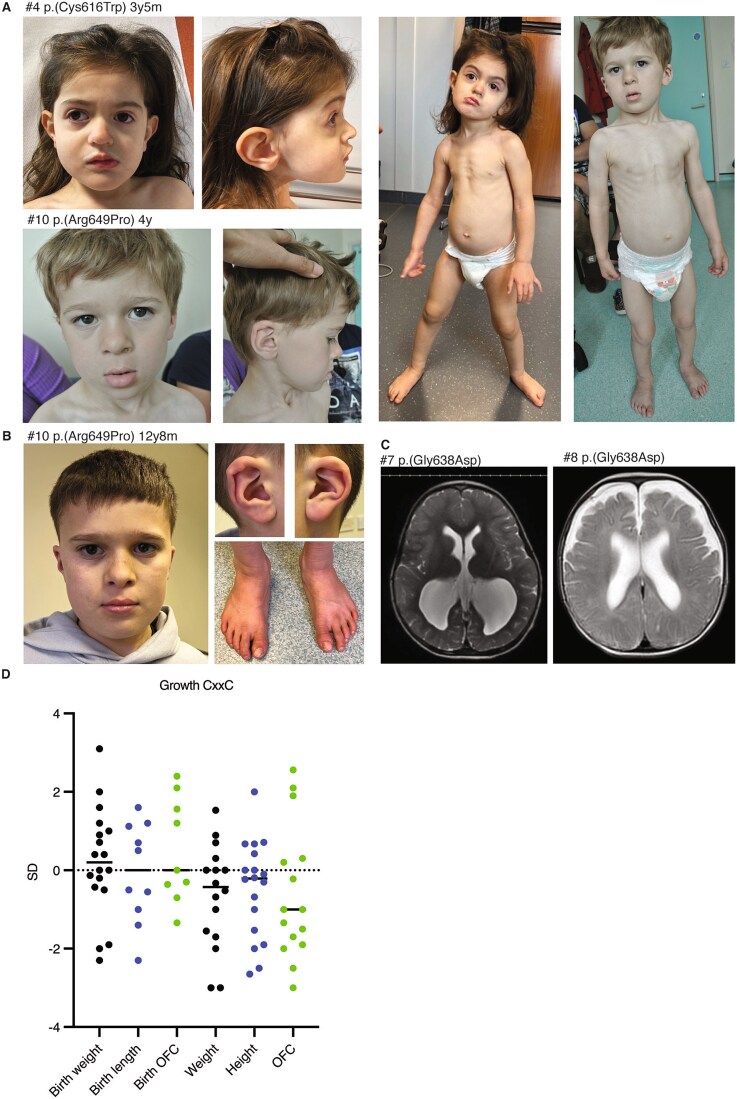
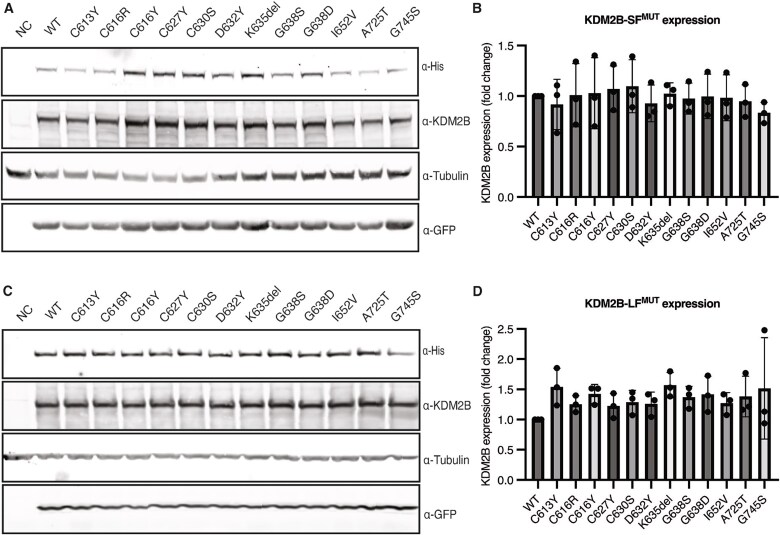
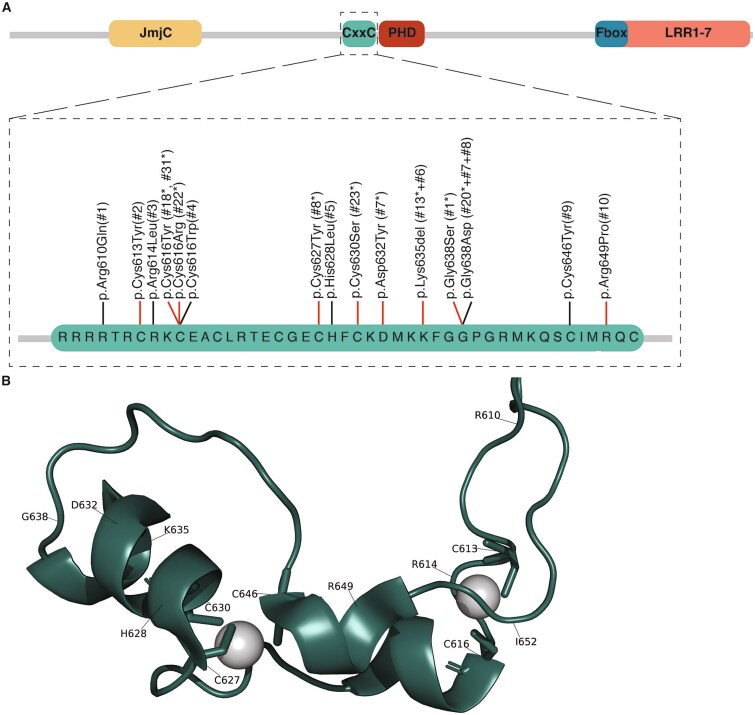
Rare variants affecting the epigenetic regulator KDM2B cause a recently delineated neurodevelopmental disorder. Interestingly, we previously identified both a general KDM2B-associated episignature and a subsignature specific to variants in the DNA-binding CxxC domain. In light of the existence of a distinct subsignature, we set out to determine if KDM2B CxxC variants are associated with a unique phenotype and disease mechanism. We recruited individuals with heterozygous CxxC variants and assessed the variants' effect on protein expression and DNA-binding ability. We analyzed clinical data from 19 individuals, including ten previously undescribed individuals with seven novel CxxC variants. The core phenotype of the KDM2B-CxxC cohort is more extensive as compared to that of individuals with KDM2B haploinsufficiency. All individuals with CxxC variants presented with developmental delay, mainly in the speech and motor domain, in addition to variable intellectual disability and mild facial dysmorphism. Congenital heart defects were observed in up to 78% of individuals, with additional common findings including musculoskeletal, ophthalmological, and urogenital anomalies, as well as behavioral challenges and feeding difficulties. Functional assays revealed that while mutant KDM2B protein with CxxC variants can be expressed in vitro, its DNA-binding ability is significantly reduced compared to wildtype. This study shows that KDM2B CxxC variants cause a distinct neurodevelopmental syndrome, possibly through a molecular mechanism different from haploinsufficiency.
期刊介绍:
Human Molecular Genetics concentrates on full-length research papers covering a wide range of topics in all aspects of human molecular genetics. These include:
the molecular basis of human genetic disease
developmental genetics
cancer genetics
neurogenetics
chromosome and genome structure and function
therapy of genetic disease
stem cells in human genetic disease and therapy, including the application of iPS cells
genome-wide association studies
mouse and other models of human diseases
functional genomics
computational genomics
In addition, the journal also publishes research on other model systems for the analysis of genes, especially when there is an obvious relevance to human genetics.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


