Stuti Bakshi, Taryn Diep, Brandon J Willis, Rachel Reyes, Grace F Wu, Georgios Makris, Martin Poms, Isabel Day, Qin Sun, Irina Zhuravka, Lindsay Lueptow, Michelle Tang, Gareth A Cromie, Aimée M Dudley, Johannes Häberle, Gerald S Lipshutz
{"title":"A hypomorphic model of CPS1 deficiency for investigating the effects of hyperammonemia on the developing nervous system.","authors":"Stuti Bakshi, Taryn Diep, Brandon J Willis, Rachel Reyes, Grace F Wu, Georgios Makris, Martin Poms, Isabel Day, Qin Sun, Irina Zhuravka, Lindsay Lueptow, Michelle Tang, Gareth A Cromie, Aimée M Dudley, Johannes Häberle, Gerald S Lipshutz","doi":"10.1242/dmm.052303","DOIUrl":null,"url":null,"abstract":"<p><p>Carbamoyl phosphate synthetase 1 (CPS1) deficiency is a rare metabolic disorder that, in neonatal onset, is typically characterized by severe life-threatening and neurologically injuring hyperammonemic episodes with high unmet patient need. Patients that retain limited enzyme activity may present later in life with less severe hyperammonemia. CPS1 drives the first step in the urea cycle, the pathway terrestrial mammals utilize to metabolize nitrogen. In order to probe the effect of hyperammonemia on the developing nervous system and explore new therapies, a murine Cps1 exon 3-4 mutant was previously generated. However, these mice die within 24 h of birth, limiting study capabilities. Herein, we developed a novel Cps1 hypomorphic murine model with residual enzyme activity that maintains survival, but with dysfunction of Cps1 that could be detected biochemically. Characterization, based on the orthologous human variant Asn674Ile, revealed that the variant is reproducible, 100% penetrant and biochemically phenocopies the human disorder. The hypomorph presents with elevated ammonia and glutamate, and reduced citrulline, and with an impaired rate of ureagenesis, providing a novel platform to study and develop therapies for CPS1 deficiency.</p>","PeriodicalId":11144,"journal":{"name":"Disease Models & Mechanisms","volume":" ","pages":""},"PeriodicalIF":3.3000,"publicationDate":"2025-07-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12208401/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Disease Models & Mechanisms","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1242/dmm.052303","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/6/20 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"CELL BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
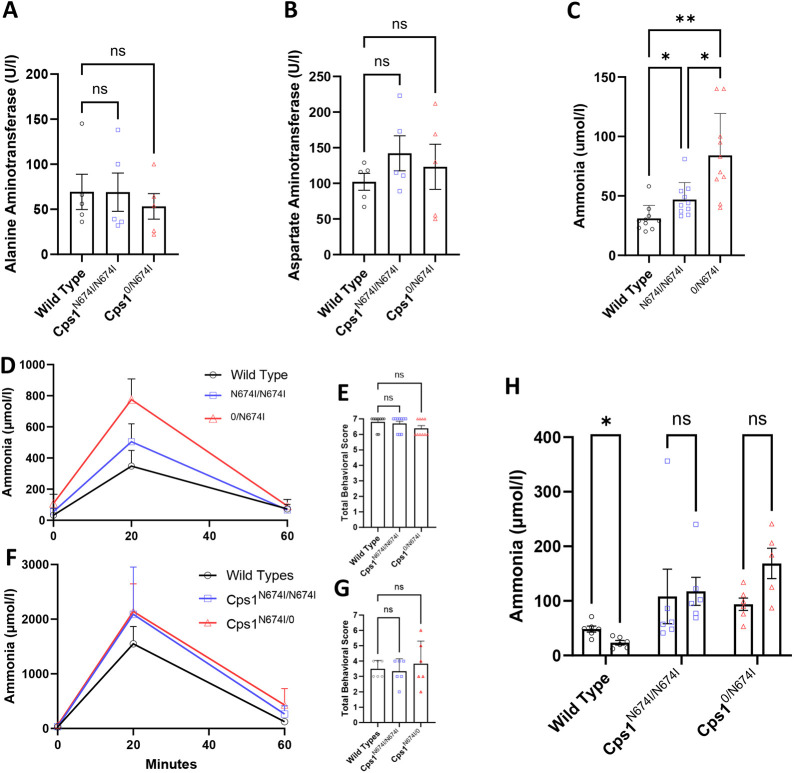
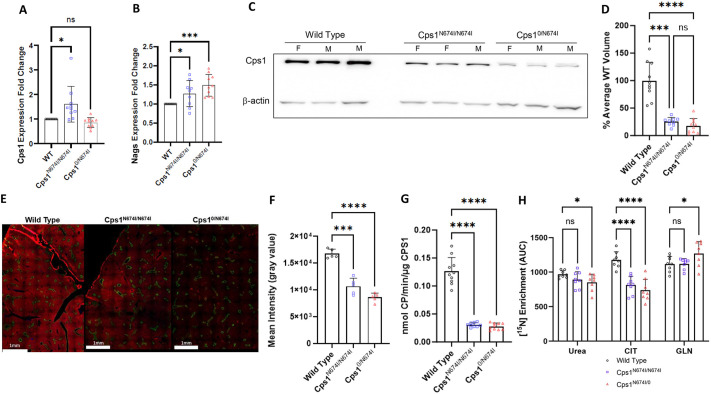
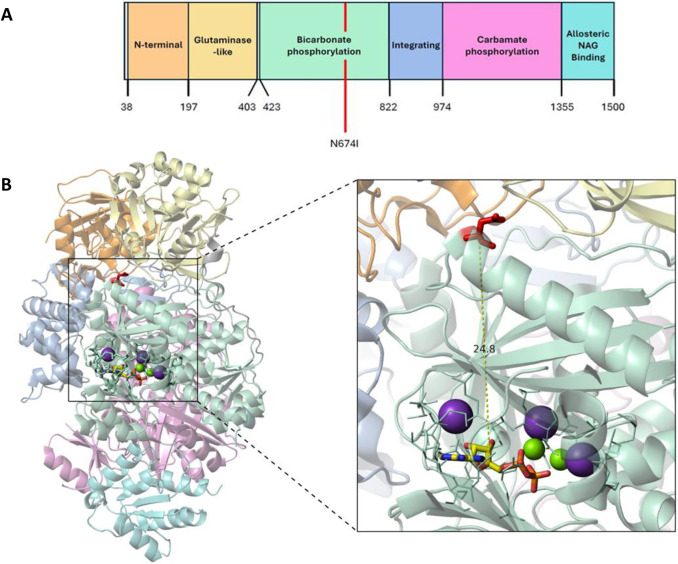
Carbamoyl phosphate synthetase 1 (CPS1) deficiency is a rare metabolic disorder that, in neonatal onset, is typically characterized by severe life-threatening and neurologically injuring hyperammonemic episodes with high unmet patient need. Patients that retain limited enzyme activity may present later in life with less severe hyperammonemia. CPS1 drives the first step in the urea cycle, the pathway terrestrial mammals utilize to metabolize nitrogen. In order to probe the effect of hyperammonemia on the developing nervous system and explore new therapies, a murine Cps1 exon 3-4 mutant was previously generated. However, these mice die within 24 h of birth, limiting study capabilities. Herein, we developed a novel Cps1 hypomorphic murine model with residual enzyme activity that maintains survival, but with dysfunction of Cps1 that could be detected biochemically. Characterization, based on the orthologous human variant Asn674Ile, revealed that the variant is reproducible, 100% penetrant and biochemically phenocopies the human disorder. The hypomorph presents with elevated ammonia and glutamate, and reduced citrulline, and with an impaired rate of ureagenesis, providing a novel platform to study and develop therapies for CPS1 deficiency.
期刊介绍:
Disease Models & Mechanisms (DMM) is an online Open Access journal focusing on the use of model systems to better understand, diagnose and treat human disease.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


