Chengwei Yang, Chao Yang, Yunxia Liang, Hongxia Yan, Aodi Zhang, Guixian Ge, Wentao Wang and Pengfei Ou
{"title":"Machine-learning enables nitrogen reduction reaction on transition metal doped C3B by controlling the charge states†","authors":"Chengwei Yang, Chao Yang, Yunxia Liang, Hongxia Yan, Aodi Zhang, Guixian Ge, Wentao Wang and Pengfei Ou","doi":"10.1039/D5QM00140D","DOIUrl":null,"url":null,"abstract":"<p >Transition metal (TM)-doped monolayer semiconductors have attracted significant attention as electrocatalysts for various applications. However, conventional density functional theory calculations often yield inaccurate predictions due to the omission of charge states, due to which extensive efforts to explore promising electrocatalysts are in vain. Here, we report a computational pipeline for high-throughput screening that combines charge-state-aware DFT calculations for stability and activity predictions with machine learning (ML)-enabled feature and mechanism analysis. Applying this pipeline to a TM-doped C<small><sub>3</sub></small>B monolayer (TM@C<small><sub>3</sub></small>B) to search for potential nitrogen reduction reaction (NRR) electrocatalysts, we initially identified 92 types of stable charge states of TM@C<small><sub>3</sub></small>B under B-rich conditions. By considering both activity and selectivity, we identified V<small><sub>C</sub></small>@C<small><sub>3</sub></small>B (V-doped at the C site in either the 0 or +1 charge state) as a promising candidate, which exhibited both low limiting potentials and excellent selectivity for the NRR. Further ML analysis of the N<small><sub>2</sub></small> adsorption energy and the first and last hydrogenation steps of TM@C<small><sub>3</sub></small>B revealed that charge transfer and the d-band center are critical factors governing NRR performance, both of which can be modulated by the different charge states. This study highlights the necessity of charge state calculations in electrochemical reaction modeling, paving a new pathway for the rational design of high-performance NRR electrocatalysts.</p>","PeriodicalId":86,"journal":{"name":"Materials Chemistry Frontiers","volume":" 11","pages":" 1681-1689"},"PeriodicalIF":6.4000,"publicationDate":"2025-04-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Materials Chemistry Frontiers","FirstCategoryId":"88","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2025/qm/d5qm00140d","RegionNum":2,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
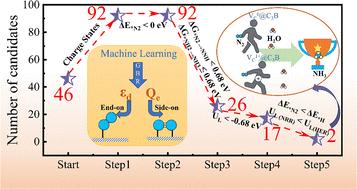
Transition metal (TM)-doped monolayer semiconductors have attracted significant attention as electrocatalysts for various applications. However, conventional density functional theory calculations often yield inaccurate predictions due to the omission of charge states, due to which extensive efforts to explore promising electrocatalysts are in vain. Here, we report a computational pipeline for high-throughput screening that combines charge-state-aware DFT calculations for stability and activity predictions with machine learning (ML)-enabled feature and mechanism analysis. Applying this pipeline to a TM-doped C3B monolayer (TM@C3B) to search for potential nitrogen reduction reaction (NRR) electrocatalysts, we initially identified 92 types of stable charge states of TM@C3B under B-rich conditions. By considering both activity and selectivity, we identified VC@C3B (V-doped at the C site in either the 0 or +1 charge state) as a promising candidate, which exhibited both low limiting potentials and excellent selectivity for the NRR. Further ML analysis of the N2 adsorption energy and the first and last hydrogenation steps of TM@C3B revealed that charge transfer and the d-band center are critical factors governing NRR performance, both of which can be modulated by the different charge states. This study highlights the necessity of charge state calculations in electrochemical reaction modeling, paving a new pathway for the rational design of high-performance NRR electrocatalysts.
期刊介绍:
Materials Chemistry Frontiers focuses on the synthesis and chemistry of exciting new materials, and the development of improved fabrication techniques. Characterisation and fundamental studies that are of broad appeal are also welcome.
This is the ideal home for studies of a significant nature that further the development of organic, inorganic, composite and nano-materials.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


