A Chevais, N V Tarbaeva, Y Y Golubkina, M M Gadzhimuradova, K V Ivashchenko, D O Ladygina, M V Vorontsova, O B Bezlepkina, G A Melnichenko, N G Mokrysheva
{"title":"[Difficulty with differential diagnosis on adrenal lesions in congenital adrenal cortex dysfunction: a series of clinical cases].","authors":"A Chevais, N V Tarbaeva, Y Y Golubkina, M M Gadzhimuradova, K V Ivashchenko, D O Ladygina, M V Vorontsova, O B Bezlepkina, G A Melnichenko, N G Mokrysheva","doi":"10.14341/probl13564","DOIUrl":null,"url":null,"abstract":"<p><p>Congenital adrenal hyperplasia (CAH) encompasses a group of autosomal recessive disorders characterized by defects in enzymes critical for steroidogenesis, with 21-hydroxylase deficiency due to mutations in the CYP21A2 gene being the most prevalent form.Since the introduction of replacement therapy and neonatal screening programs in the 1950s, there has been a significant increase in survival rates among newborns diagnosed with CAH. However, despite these advancements, mortality associated with this condition remains disproportionately high. Achieving optimal therapeutic compensation through medication remains a complex challenge, contributing to a range of long-term complications. These complications stem from both the underlying disease and its treatment, impacting key physiological functions, including metabolism, growth and development, cardiovascular health, and fertility. These multifaceted outcomes underscore the need for ongoing research and the refinement of therapeutic approaches to better manage this intricate condition. This article presents a series of four clinical cases of CAH characterized by the absence of sustained compensation for glucoand mineralocorticoid deficiencies. These cases were further complicated by the development of large adrenal masses and ectopic testicular adrenal rest tissue (TART), emphasizing the challenges in achieving long-term disease management.</p>","PeriodicalId":101419,"journal":{"name":"Problemy endokrinologii","volume":"71 2","pages":"22-34"},"PeriodicalIF":0.0000,"publicationDate":"2025-05-20","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12117987/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Problemy endokrinologii","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.14341/probl13564","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
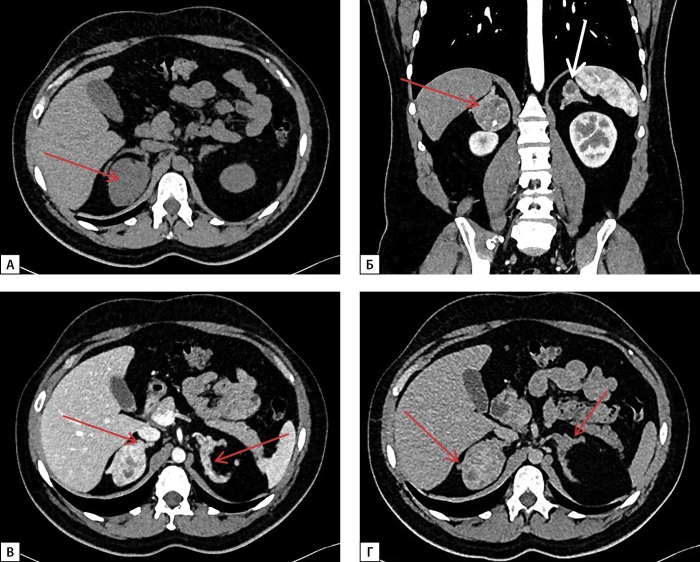
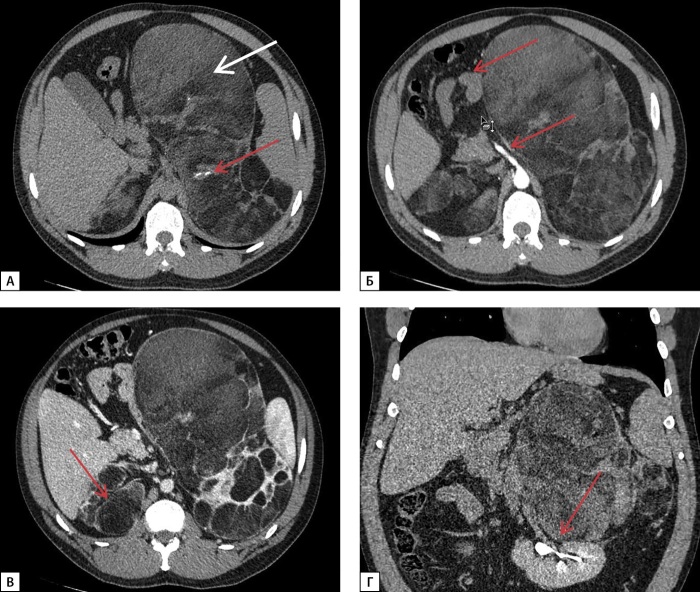
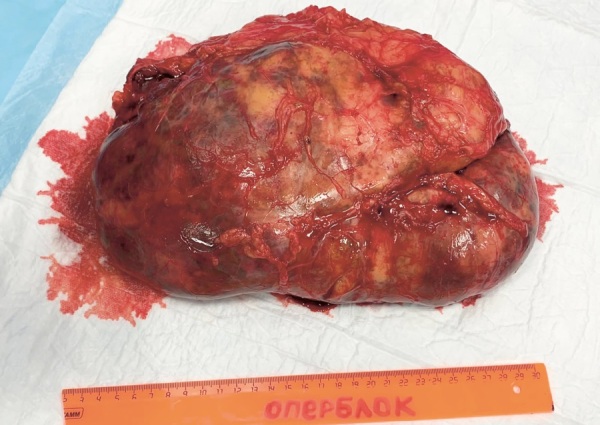
Congenital adrenal hyperplasia (CAH) encompasses a group of autosomal recessive disorders characterized by defects in enzymes critical for steroidogenesis, with 21-hydroxylase deficiency due to mutations in the CYP21A2 gene being the most prevalent form.Since the introduction of replacement therapy and neonatal screening programs in the 1950s, there has been a significant increase in survival rates among newborns diagnosed with CAH. However, despite these advancements, mortality associated with this condition remains disproportionately high. Achieving optimal therapeutic compensation through medication remains a complex challenge, contributing to a range of long-term complications. These complications stem from both the underlying disease and its treatment, impacting key physiological functions, including metabolism, growth and development, cardiovascular health, and fertility. These multifaceted outcomes underscore the need for ongoing research and the refinement of therapeutic approaches to better manage this intricate condition. This article presents a series of four clinical cases of CAH characterized by the absence of sustained compensation for glucoand mineralocorticoid deficiencies. These cases were further complicated by the development of large adrenal masses and ectopic testicular adrenal rest tissue (TART), emphasizing the challenges in achieving long-term disease management.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


