Neblina Sikta, Samuel Gooley, Timothy E Green, Olivia Hoeper, Tom Witkowski, Caitlin Bennett, David Francis, Joshua Reid, Kevin Mao, Mohammed Awad, Samuel Roberts-Thomson, Kristian Bulluss, Jonathan Clark, Ingrid E Scheffer, Piero Perucca, Mark F Bennett, Melanie Bahlo, Samuel F Berkovic, Michael S Hildebrand
{"title":"Improving genetic diagnostic yield in familial and sporadic cerebral cavernous malformations: detection of copy number and deep Intronic variants.","authors":"Neblina Sikta, Samuel Gooley, Timothy E Green, Olivia Hoeper, Tom Witkowski, Caitlin Bennett, David Francis, Joshua Reid, Kevin Mao, Mohammed Awad, Samuel Roberts-Thomson, Kristian Bulluss, Jonathan Clark, Ingrid E Scheffer, Piero Perucca, Mark F Bennett, Melanie Bahlo, Samuel F Berkovic, Michael S Hildebrand","doi":"10.1093/hmg/ddaf077","DOIUrl":null,"url":null,"abstract":"<p><p>Cerebral cavernous malformations (CCMs) are intracranial vascular lesions associated with risk of haemorrhages and seizures. While the majority are sporadic and often associated with somatic variants in PIK3CA and MAP3K3, around 20% are familial with germline variants in one of three CCM genes-KRIT1/CCM1, CCM2 and PDCD10/CCM3. We performed comprehensive phenotyping and genetic analysis of nine multiplex families and ten sporadic individuals with CCM. In the familial cases, initial standard analyses had a low yield, we therefore searched for small copy number changes and deep intronic variants. Subsequently, pathogenic germline variants in KRIT1/CCM1 or CCM2 were identified in all 9 multiplex families. Single or multiple exon deletions or splice site variants in KRIT1/CCM1 were found in 3/9 families. Where cavernous malformation tissue was available, second hit somatic PIK3CA variants were identified in 4/7 individuals. These 4 individuals were from separate families with germline KRIT1/CCM1 variants. In 8/10 sporadic cases, we detected recurrent pathogenic somatic PIK3CA, MAP3K3 or CCM2 variants. All familial cases had multiple CCMs, whereas the sporadic cases had a single lesion only, which was in the temporal lobe in 9/10 individuals. Our comprehensive approach interrogating deep intronic variants combined with detection of small copy number variants warrants implementation in standard clinical genetic testing pipelines to increase diagnostic yield. We also build on the established second hit germline and somatic variant mechanism in some CCM lesions. Genetic diagnosis has clinical implications such as reproductive counselling and provides potential eligibility for precision medicine therapies to treat rapidly growing CCMs.</p>","PeriodicalId":13070,"journal":{"name":"Human molecular genetics","volume":" ","pages":"1286-1293"},"PeriodicalIF":3.2000,"publicationDate":"2025-07-20","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12278727/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Human molecular genetics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1093/hmg/ddaf077","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
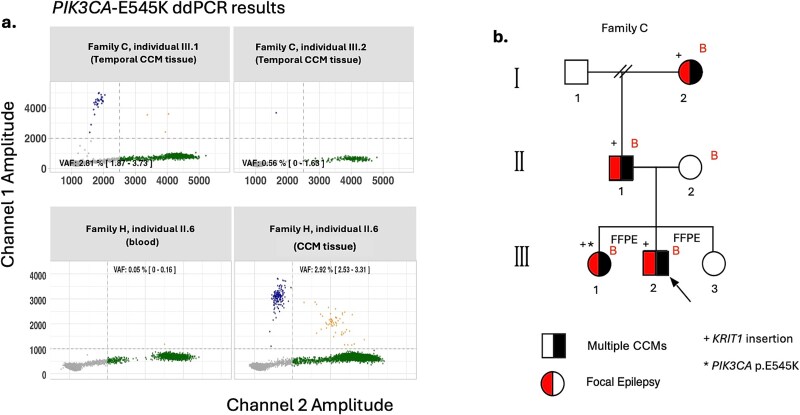
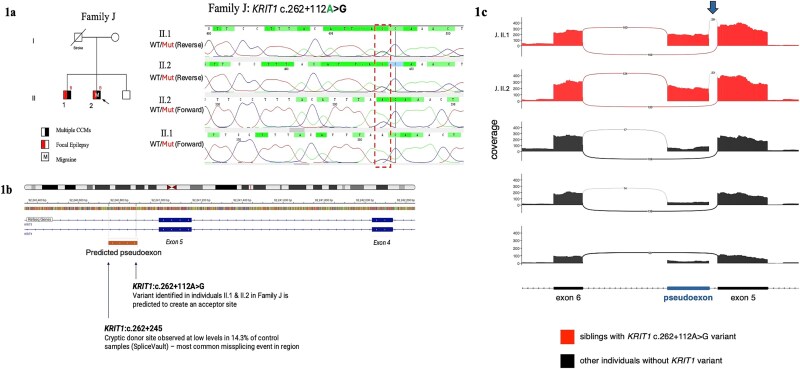
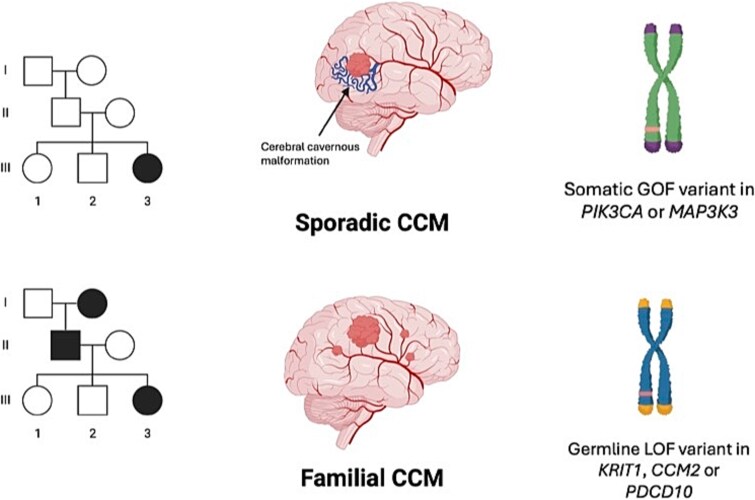
Cerebral cavernous malformations (CCMs) are intracranial vascular lesions associated with risk of haemorrhages and seizures. While the majority are sporadic and often associated with somatic variants in PIK3CA and MAP3K3, around 20% are familial with germline variants in one of three CCM genes-KRIT1/CCM1, CCM2 and PDCD10/CCM3. We performed comprehensive phenotyping and genetic analysis of nine multiplex families and ten sporadic individuals with CCM. In the familial cases, initial standard analyses had a low yield, we therefore searched for small copy number changes and deep intronic variants. Subsequently, pathogenic germline variants in KRIT1/CCM1 or CCM2 were identified in all 9 multiplex families. Single or multiple exon deletions or splice site variants in KRIT1/CCM1 were found in 3/9 families. Where cavernous malformation tissue was available, second hit somatic PIK3CA variants were identified in 4/7 individuals. These 4 individuals were from separate families with germline KRIT1/CCM1 variants. In 8/10 sporadic cases, we detected recurrent pathogenic somatic PIK3CA, MAP3K3 or CCM2 variants. All familial cases had multiple CCMs, whereas the sporadic cases had a single lesion only, which was in the temporal lobe in 9/10 individuals. Our comprehensive approach interrogating deep intronic variants combined with detection of small copy number variants warrants implementation in standard clinical genetic testing pipelines to increase diagnostic yield. We also build on the established second hit germline and somatic variant mechanism in some CCM lesions. Genetic diagnosis has clinical implications such as reproductive counselling and provides potential eligibility for precision medicine therapies to treat rapidly growing CCMs.
期刊介绍:
Human Molecular Genetics concentrates on full-length research papers covering a wide range of topics in all aspects of human molecular genetics. These include:
the molecular basis of human genetic disease
developmental genetics
cancer genetics
neurogenetics
chromosome and genome structure and function
therapy of genetic disease
stem cells in human genetic disease and therapy, including the application of iPS cells
genome-wide association studies
mouse and other models of human diseases
functional genomics
computational genomics
In addition, the journal also publishes research on other model systems for the analysis of genes, especially when there is an obvious relevance to human genetics.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


