Robin Ketteler, Koshiro Kiso, Lucas von Chamier, Alexander Agrotis
{"title":"ATG5 is dispensable for ATG8ylation of cellular proteins.","authors":"Robin Ketteler, Koshiro Kiso, Lucas von Chamier, Alexander Agrotis","doi":"10.1080/27694127.2024.2392450","DOIUrl":null,"url":null,"abstract":"<p><p>Protein ATG8ylation refers to a post-translational modification involving covalent attachment of ubiquitin-like autophagy-related protein ATG8 (LC3/GABARAP) to other cellular proteins, with reversal mediated by ATG4 proteases. While lipid ATG8ylation is important for autophagosome formation and mechanistically well-characterized, little is known about the mechanism of protein ATG8ylation. Here, we investigated the conjugation machinery of protein ATG8ylation in CRISPR/Cas9-engineered knockout human cell lines, utilizing a deconjugation-resistant (Q116P G120) form of MAP1LC3B. We report that protein ATG8ylation requires the E1-like activating enzyme ATG7 and E2-like conjugating enzyme ATG3, in common with ATG8 lipidation. However, in contrast, the E3-like ATG12-ATG5-ATG16L1 complex involved in lipidation is dispensable for protein ATG8ylation, since ATG5 knockout cells can form ATG8ylated protein conjugates. Further, we uncover that ATG7 itself is a target of ATG8ylation. Overall, our work provides crucial insight into the mechanism of protein ATG8ylation, distinguishing it from ATG8 lipidation, which will aid investigating its functional role.</p>","PeriodicalId":72341,"journal":{"name":"Autophagy reports","volume":"3 1","pages":"2392450"},"PeriodicalIF":0.0000,"publicationDate":"2024-10-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11864658/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Autophagy reports","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1080/27694127.2024.2392450","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
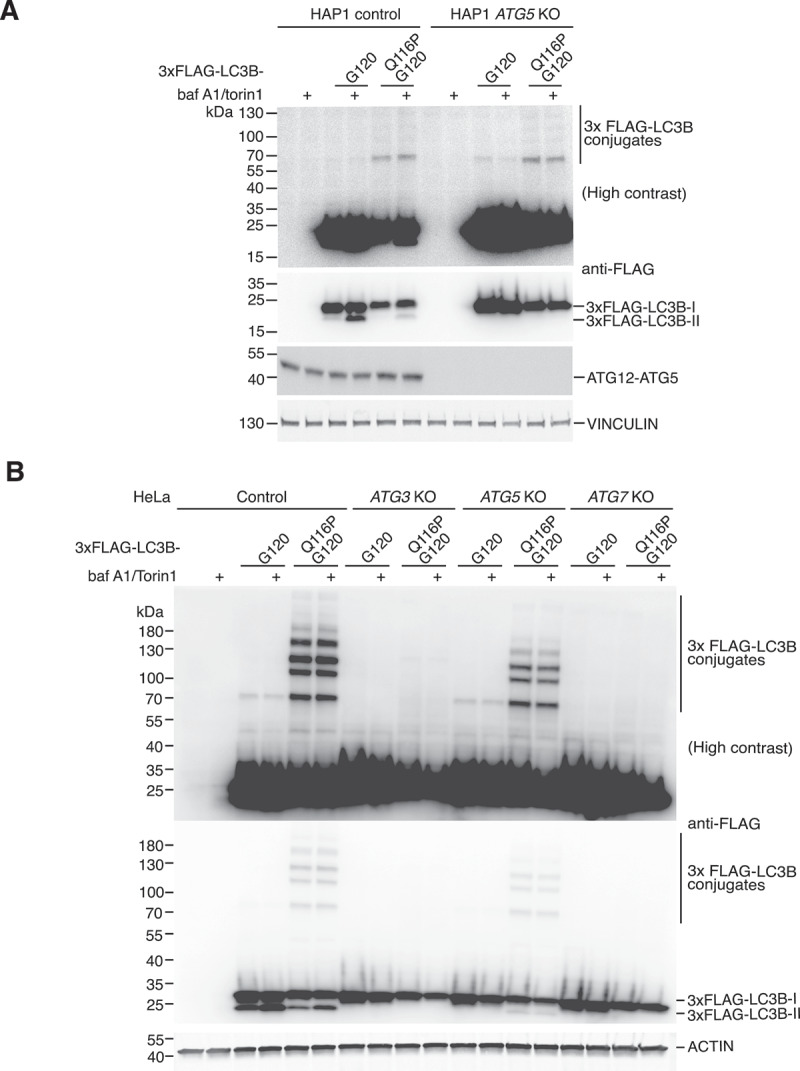
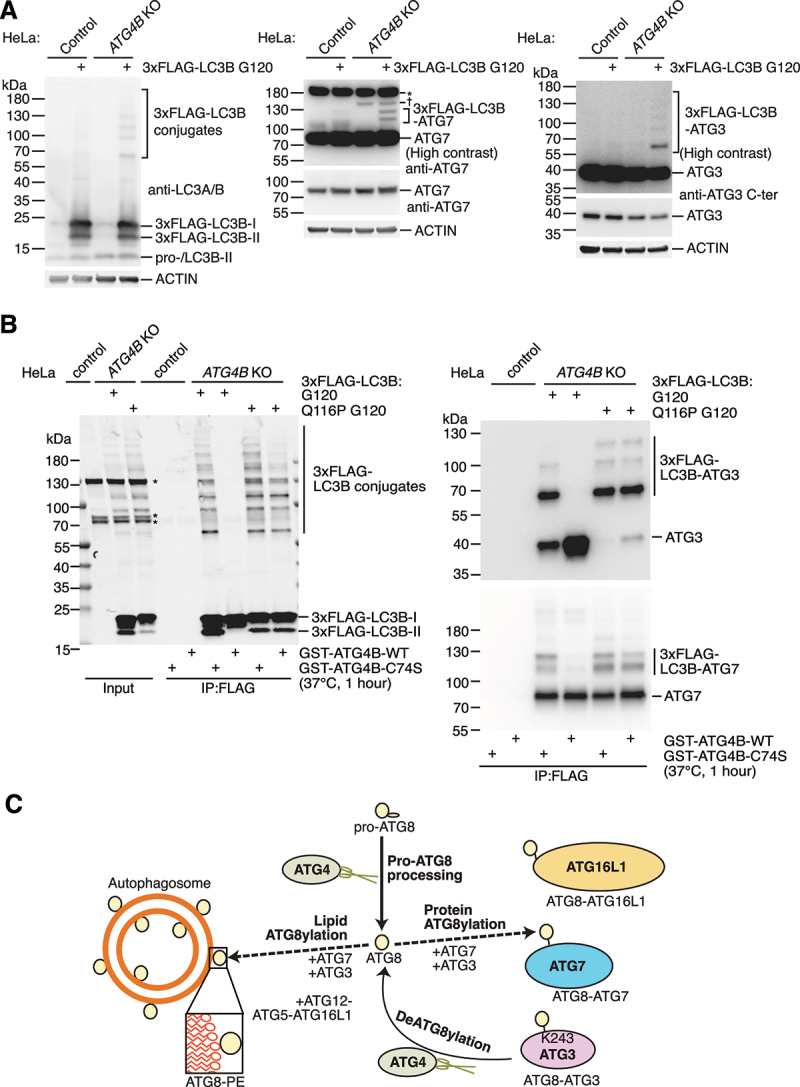
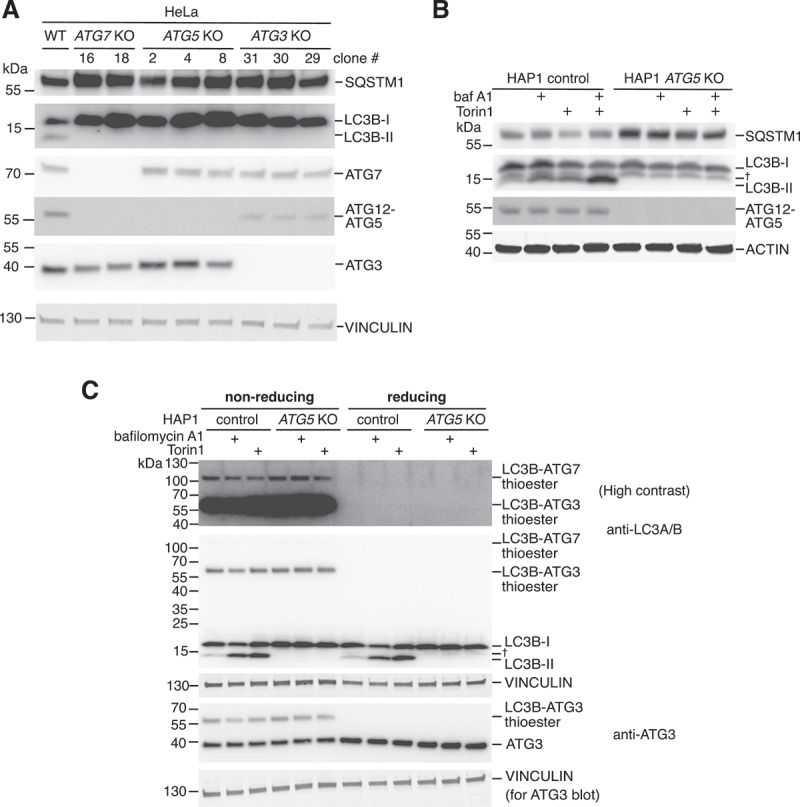
Protein ATG8ylation refers to a post-translational modification involving covalent attachment of ubiquitin-like autophagy-related protein ATG8 (LC3/GABARAP) to other cellular proteins, with reversal mediated by ATG4 proteases. While lipid ATG8ylation is important for autophagosome formation and mechanistically well-characterized, little is known about the mechanism of protein ATG8ylation. Here, we investigated the conjugation machinery of protein ATG8ylation in CRISPR/Cas9-engineered knockout human cell lines, utilizing a deconjugation-resistant (Q116P G120) form of MAP1LC3B. We report that protein ATG8ylation requires the E1-like activating enzyme ATG7 and E2-like conjugating enzyme ATG3, in common with ATG8 lipidation. However, in contrast, the E3-like ATG12-ATG5-ATG16L1 complex involved in lipidation is dispensable for protein ATG8ylation, since ATG5 knockout cells can form ATG8ylated protein conjugates. Further, we uncover that ATG7 itself is a target of ATG8ylation. Overall, our work provides crucial insight into the mechanism of protein ATG8ylation, distinguishing it from ATG8 lipidation, which will aid investigating its functional role.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


