Annemiek Maaskant, Donghyeok Lee, Huy Ngo, Roy C Montijn, Jaco Bakker, Jan A M Langermans, Evgeni Levin
{"title":"AI for rapid identification of major butyrate-producing bacteria in rhesus macaques (Macaca mulatta).","authors":"Annemiek Maaskant, Donghyeok Lee, Huy Ngo, Roy C Montijn, Jaco Bakker, Jan A M Langermans, Evgeni Levin","doi":"10.1186/s42523-025-00410-2","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>The gut microbiome plays a crucial role in health and disease, influencing digestion, metabolism, and immune function. Traditional microbiome analysis methods are often expensive, time-consuming, and require specialized expertise, limiting their practical application in clinical settings. Evolving artificial intelligence (AI) technologies present opportunities for developing alternative methods. However, the lack of transparency in these technologies limits the ability of clinicians to incorporate AI-driven diagnostic tools into their healthcare systems. The aim of this study was to investigate an AI approach that rapidly predicts different bacterial genera and bacterial groups, specifically butyrate producers, from digital images of fecal smears of rhesus macaques (Macaca mulatta). In addition, to improve transparency, we employed explainability analysis to uncover the image features influencing the model's predictions.</p><p><strong>Results: </strong>By integrating fecal image data with corresponding metagenomic sequencing information, the deep learning (DL) and machine learning (ML) algorithms successfully predicted 16 individual bacterial genera (area under the curve (AUC) > 0.7) among the 50 most abundant genera in rhesus macaques (Macaca mulatta). The model was successful in predicting functional groups, major butyrate producers (AUC 0.75) and a mixed group including fermenters and short-chain fatty acid (SCFA) producers (AUC 0.81). For both models of butyrate producers and mixed fermenters, the explainability experiments revealed no decline in the AUC when random noise was added to the images. Increased blurring led to a gradual decline in the AUC. The model's performance was robust against the impact of fecal shape from smearing, with a stable AUC maintained until patch 4 for all groups, as assessed through scrambling. No significant correlation was detected between the prediction probabilities and the total fecal weight used in the smear; r = 0.30 ± 0.3 (p > 0.1) and r = 0.04 ± 0.36 (p > 0.8) for the butyrate producers and mixed fermenters, respectively.</p><p><strong>Conclusion: </strong>Our approach demonstrated the ability to predict a wide range of clinically relevant microbial genera and microbial groups in the gut microbiome based on digital images from a fecal smear. The models proved to be robust to the smearing method, random noise and the amount of fecal matter. This study introduces a rapid, non-invasive, and cost-effective method for microbiome profiling, with potential applications in veterinary diagnostics.</p>","PeriodicalId":72201,"journal":{"name":"Animal microbiome","volume":"7 1","pages":"39"},"PeriodicalIF":4.4000,"publicationDate":"2025-04-24","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12020216/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Animal microbiome","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/s42523-025-00410-2","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MICROBIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Background: The gut microbiome plays a crucial role in health and disease, influencing digestion, metabolism, and immune function. Traditional microbiome analysis methods are often expensive, time-consuming, and require specialized expertise, limiting their practical application in clinical settings. Evolving artificial intelligence (AI) technologies present opportunities for developing alternative methods. However, the lack of transparency in these technologies limits the ability of clinicians to incorporate AI-driven diagnostic tools into their healthcare systems. The aim of this study was to investigate an AI approach that rapidly predicts different bacterial genera and bacterial groups, specifically butyrate producers, from digital images of fecal smears of rhesus macaques (Macaca mulatta). In addition, to improve transparency, we employed explainability analysis to uncover the image features influencing the model's predictions.
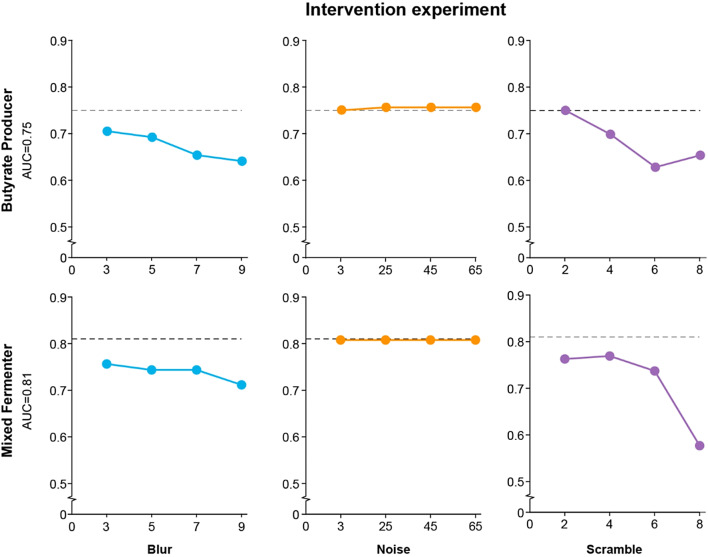
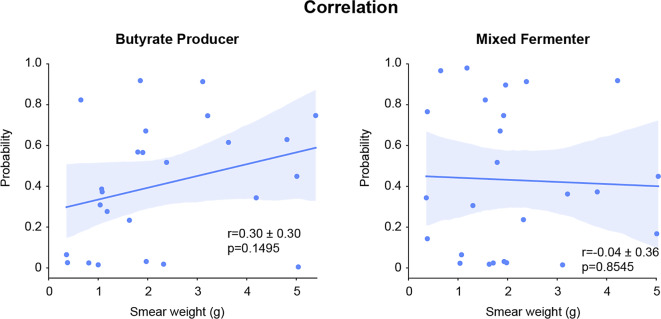
Results: By integrating fecal image data with corresponding metagenomic sequencing information, the deep learning (DL) and machine learning (ML) algorithms successfully predicted 16 individual bacterial genera (area under the curve (AUC) > 0.7) among the 50 most abundant genera in rhesus macaques (Macaca mulatta). The model was successful in predicting functional groups, major butyrate producers (AUC 0.75) and a mixed group including fermenters and short-chain fatty acid (SCFA) producers (AUC 0.81). For both models of butyrate producers and mixed fermenters, the explainability experiments revealed no decline in the AUC when random noise was added to the images. Increased blurring led to a gradual decline in the AUC. The model's performance was robust against the impact of fecal shape from smearing, with a stable AUC maintained until patch 4 for all groups, as assessed through scrambling. No significant correlation was detected between the prediction probabilities and the total fecal weight used in the smear; r = 0.30 ± 0.3 (p > 0.1) and r = 0.04 ± 0.36 (p > 0.8) for the butyrate producers and mixed fermenters, respectively.
Conclusion: Our approach demonstrated the ability to predict a wide range of clinically relevant microbial genera and microbial groups in the gut microbiome based on digital images from a fecal smear. The models proved to be robust to the smearing method, random noise and the amount of fecal matter. This study introduces a rapid, non-invasive, and cost-effective method for microbiome profiling, with potential applications in veterinary diagnostics.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


