{"title":"Efficiently constructing complete genomes with CycloneSEQ to fill gaps in bacterial draft assemblies.","authors":"Hewei Liang, Yuanqiang Zou, Mengmeng Wang, Tongyuan Hu, Haoyu Wang, Wenxin He, Yanmei Ju, Ruijin Guo, Junyi Chen, Fei Guo, Tao Zeng, Yuliang Dong, Yuning Zhang, Bo Wang, Chuanyu Liu, Xin Jin, Wenwei Zhang, Xun Xu, Liang Xiao","doi":"10.46471/gigabyte.154","DOIUrl":null,"url":null,"abstract":"<p><p>Current microbial sequencing relies on short-read platforms like Illumina and DNBSEQ, which are cost-effective and accurate but often produce fragmented draft genomes. Here, we used CycloneSEQ for long-read sequencing of ATCC BAA-835, producing long-reads with an average length of 11.6 kbp and an average quality score of 14.4. Hybrid assembly with short-reads data resulted in an error rate of only 0.04 mismatches and 0.08 indels per 100 kbp compared to the reference genome. This method, validated across nine species, successfully assembled complete circular genomes. Hybrid assembly significantly enhances genome completeness by using long-reads to fill gaps and accurately assembling multi-copy rRNA genes, unlike short-reads alone. Data subsampling showed that combining over 500 Mbp of short-read data with 100 Mbp of long-read data yields high-quality circular assemblies. CycloneSEQ long-reads improves the assembly of circular complete genomes from mixed microbial communities; however, its base quality needs improving. Integrating DNBSEQ short-reads improved accuracy, resulting in complete and accurate assemblies.</p>","PeriodicalId":73157,"journal":{"name":"GigaByte (Hong Kong, China)","volume":"2025 ","pages":"gigabyte154"},"PeriodicalIF":1.2000,"publicationDate":"2025-04-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12051259/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"GigaByte (Hong Kong, China)","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.46471/gigabyte.154","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
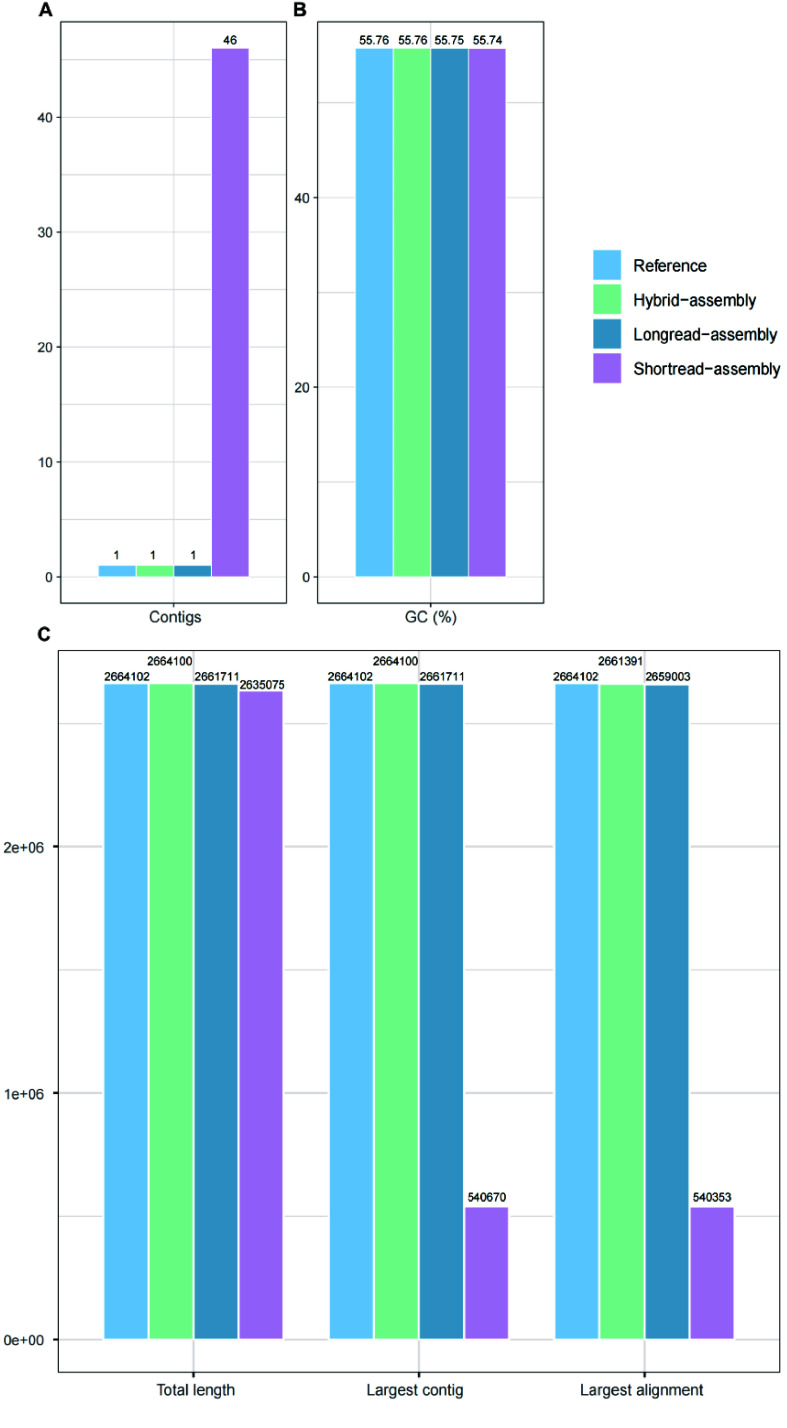
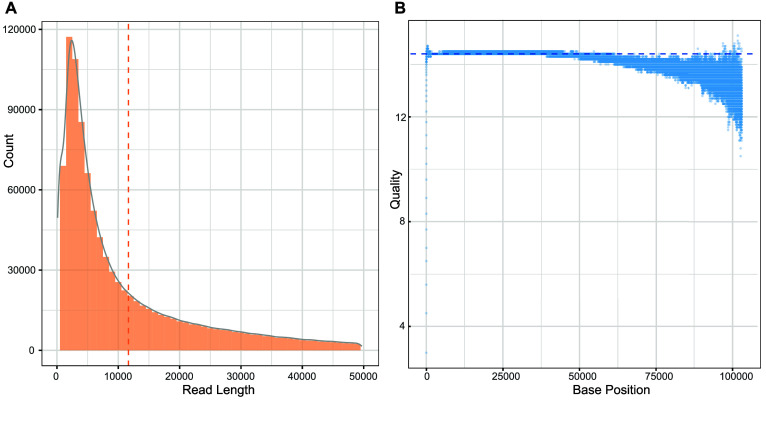
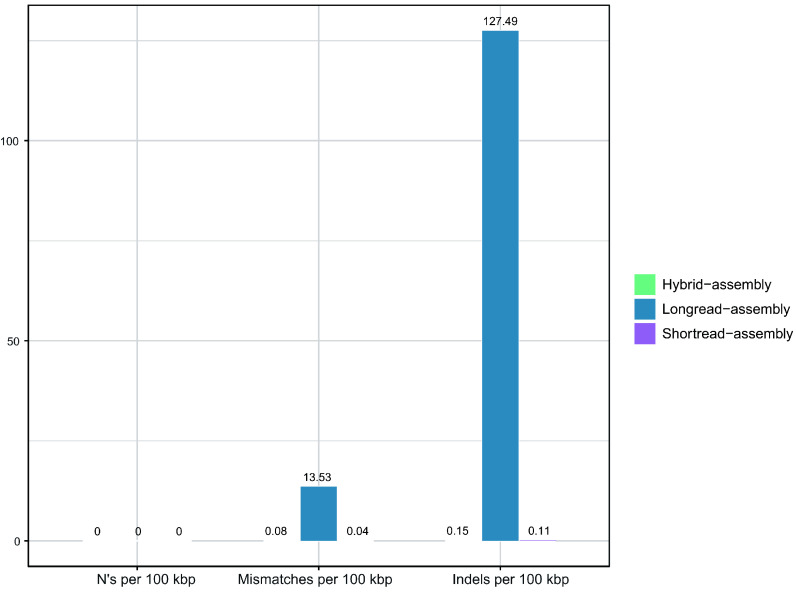
Current microbial sequencing relies on short-read platforms like Illumina and DNBSEQ, which are cost-effective and accurate but often produce fragmented draft genomes. Here, we used CycloneSEQ for long-read sequencing of ATCC BAA-835, producing long-reads with an average length of 11.6 kbp and an average quality score of 14.4. Hybrid assembly with short-reads data resulted in an error rate of only 0.04 mismatches and 0.08 indels per 100 kbp compared to the reference genome. This method, validated across nine species, successfully assembled complete circular genomes. Hybrid assembly significantly enhances genome completeness by using long-reads to fill gaps and accurately assembling multi-copy rRNA genes, unlike short-reads alone. Data subsampling showed that combining over 500 Mbp of short-read data with 100 Mbp of long-read data yields high-quality circular assemblies. CycloneSEQ long-reads improves the assembly of circular complete genomes from mixed microbial communities; however, its base quality needs improving. Integrating DNBSEQ short-reads improved accuracy, resulting in complete and accurate assemblies.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


