A random forest-based predictive model for classifying BRCA1 missense variants: a novel approach for evaluating the missense mutations effect
IF 2.5
3区 生物学
Q2 GENETICS & HEREDITY
引用次数: 0
Abstract
The right classification of variants is the key to pre-symptomatic detection of disease and conducting preventive actions. Since BRCA1 has a high incidence and penetrance in breast and ovarian cancers, a high-performance predictive tool can be employed to classify the clinical significance of its variants. Several tools have previously been developed for this purpose which poorly classify the significance in specific cases. The proposed tools commonly assign a score without providing any interpretation behind it. To reach an accurate predictive tool with interpretation abilities, in this study, we propose BRCA1-Forest which works based on random forest as a well-known machine learning technique for making interpretable decisions with high specificity and sensitivity in variants classification. The method involves narrowing down available options until reaching the final decision. To this end, a set of BRCA1 benign and pathogenic missense variants was collected first, and then, the dataset was prepared based on the effect of each variant on the protein sequence. The dataset was enriched by adding physicochemical changes and the conservation score of the amino acid position as pathogenicity criteria. The proposed model was trained based on the dataset to classify the clinical significance of variants. The performance of BRCA1-Forest was compared to four state-of-the-art methods, SIFT, PolyPhen2, CADD, and DANN, in terms of different evaluation metrics including precision, recall, false positive rate (FPR), the area under the receiver operator curve (AUC ROC), the area under the precision-recall curve (AUC-PR), and Mathew correlation coefficient (MCC). The results reveal that the proposed model outperforms the abovementioned tools in all metrics except for recall. The software of BRCA1-Forest is available at https://github.com/HamedKAAC/BRCA1Forest .
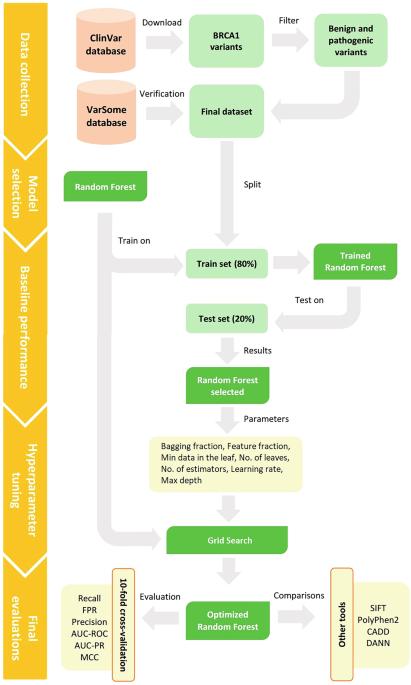
基于随机森林的BRCA1错义变异分类预测模型:一种评估错义突变效应的新方法。
正确分类变异是症状前发现疾病并采取预防行动的关键。由于BRCA1在乳腺癌和卵巢癌中具有较高的发病率和外显率,因此可以采用高性能的预测工具对其变体的临床意义进行分类。以前为此目的开发的一些工具对具体情况的重要性分类很差。建议的工具通常分配分数而不提供背后的任何解释。为了获得具有解释能力的准确预测工具,在本研究中,我们提出了基于随机森林的BRCA1-Forest作为一种众所周知的机器学习技术,在变体分类中做出具有高特异性和敏感性的可解释决策。该方法包括缩小可用选项,直到做出最终决定。为此,首先收集一组BRCA1良性和致病性错义变异体,然后根据各变异体对蛋白质序列的影响,建立数据集。通过添加理化变化和氨基酸位置的保存评分作为致病性标准,丰富了数据集。该模型基于数据集进行训练,对变异的临床意义进行分类。在精密度、召回率、假阳性率(FPR)、接收算子曲线下面积(AUC- ROC)、精确召回率曲线下面积(AUC- pr)和马修相关系数(MCC)等不同评价指标上,比较了BRCA1-Forest与SIFT、PolyPhen2、CADD和DANN四种最先进的方法的性能。结果表明,除了召回率之外,所提出的模型在所有指标上都优于上述工具。BRCA1-Forest的软件可在https://github.com/HamedKAAC/BRCA1Forest上获得。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Journal of Human Genetics
生物-遗传学
CiteScore
7.20
自引率
0.00%
发文量
101
审稿时长
4-8 weeks
期刊介绍:
The Journal of Human Genetics is an international journal publishing articles on human genetics, including medical genetics and human genome analysis. It covers all aspects of human genetics, including molecular genetics, clinical genetics, behavioral genetics, immunogenetics, pharmacogenomics, population genetics, functional genomics, epigenetics, genetic counseling and gene therapy.
Articles on the following areas are especially welcome: genetic factors of monogenic and complex disorders, genome-wide association studies, genetic epidemiology, cancer genetics, personal genomics, genotype-phenotype relationships and genome diversity.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


