Erika Yazawa, Erin M Keating, Suya Wang, Mason E Sweat, Qing Ma, Yang Xu, Michael Schlame, William T Pu
{"title":"A murine model of Barth syndrome recapitulates human cardiac and skeletal muscle phenotypes.","authors":"Erika Yazawa, Erin M Keating, Suya Wang, Mason E Sweat, Qing Ma, Yang Xu, Michael Schlame, William T Pu","doi":"10.1242/dmm.052077","DOIUrl":null,"url":null,"abstract":"<p><p>Barth syndrome is a mitochondrial disorder with hallmarks of cardiac and skeletal muscle weakness. It is caused by pathogenic variants in the X-linked gene tafazzin (TAZ), required for cardiolipin remodeling. Previously described germline and conditional Taz knockout models are not ideal for therapeutic development because they lack the combination of robust survival to adulthood, cardiomyopathy and skeletal muscle weakness. We characterized a cardiac and skeletal muscle-specific Taz knockout model (TazmKO) in which Cre recombinase is expressed from the muscle creatine kinase promoter (mCK-Cre). TazmKO mice survived normally. Cardiolipin composition was abnormal in both heart and skeletal muscle. TazmKO had reduced heart function by 2 months of age, and function progressively declined thereafter. Reduced treadmill endurance and diminished peak oxygen consumption were evident by 3 months of age, suggesting reduced skeletal muscle function. Electron microscopy showed abnormalities in mitochondrial structure and distribution. Overall, TazmKO mice display diminished cardiac function and exercise capacity while maintaining normal survival. This model will be useful for studying the effects of TAZ deficiency in striated muscles and for testing potential therapies for Barth syndrome.</p>","PeriodicalId":11144,"journal":{"name":"Disease Models & Mechanisms","volume":" ","pages":""},"PeriodicalIF":3.3000,"publicationDate":"2025-05-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12128220/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Disease Models & Mechanisms","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1242/dmm.052077","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/5/19 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"CELL BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
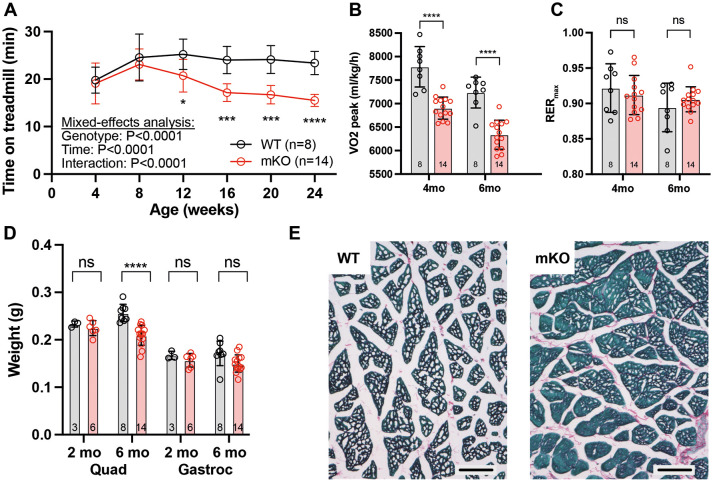
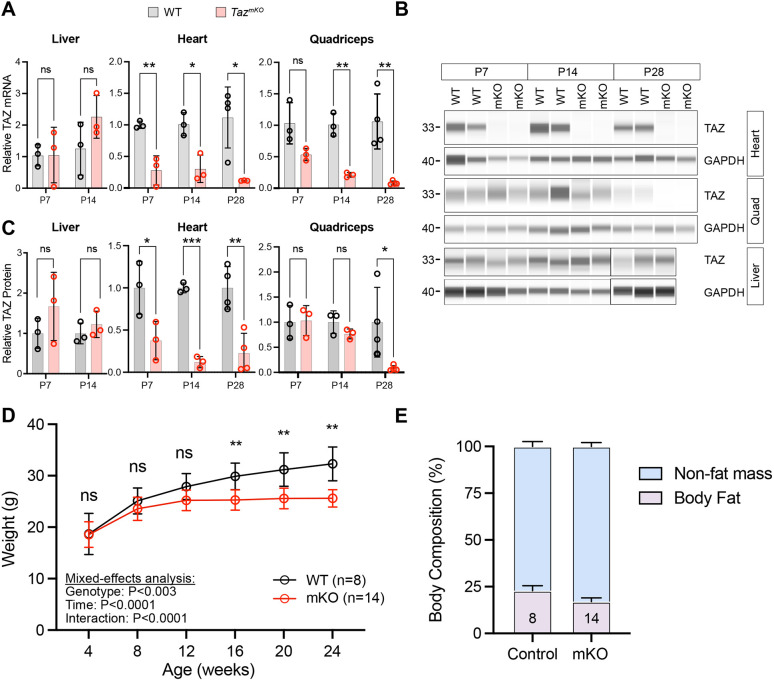
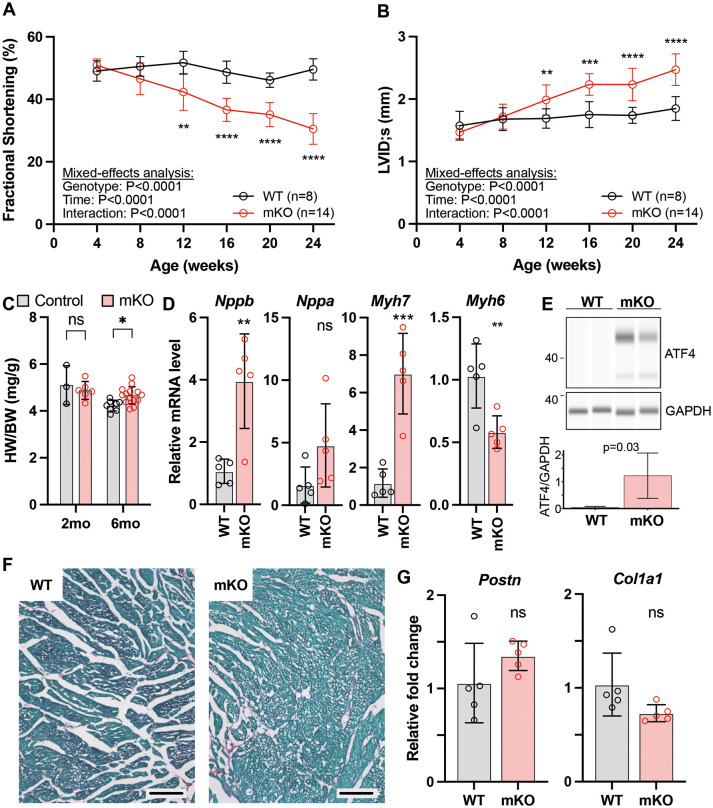
Barth syndrome is a mitochondrial disorder with hallmarks of cardiac and skeletal muscle weakness. It is caused by pathogenic variants in the X-linked gene tafazzin (TAZ), required for cardiolipin remodeling. Previously described germline and conditional Taz knockout models are not ideal for therapeutic development because they lack the combination of robust survival to adulthood, cardiomyopathy and skeletal muscle weakness. We characterized a cardiac and skeletal muscle-specific Taz knockout model (TazmKO) in which Cre recombinase is expressed from the muscle creatine kinase promoter (mCK-Cre). TazmKO mice survived normally. Cardiolipin composition was abnormal in both heart and skeletal muscle. TazmKO had reduced heart function by 2 months of age, and function progressively declined thereafter. Reduced treadmill endurance and diminished peak oxygen consumption were evident by 3 months of age, suggesting reduced skeletal muscle function. Electron microscopy showed abnormalities in mitochondrial structure and distribution. Overall, TazmKO mice display diminished cardiac function and exercise capacity while maintaining normal survival. This model will be useful for studying the effects of TAZ deficiency in striated muscles and for testing potential therapies for Barth syndrome.
期刊介绍:
Disease Models & Mechanisms (DMM) is an online Open Access journal focusing on the use of model systems to better understand, diagnose and treat human disease.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


