Alexandra Nitoiu, Qihong Zhang, Erika Tavares, Janice Min Li, Kashif Ahmed, Kit Green-Sanderson, Mahnoor Rashid, Shahir M Morcos, Jayson T Maynes, Eric I Campos, Val C Sheffield, Ajoy Vincent, Elise Héon
{"title":"Defective IFT57 underlies a novel cause of Bardet-Biedl syndrome.","authors":"Alexandra Nitoiu, Qihong Zhang, Erika Tavares, Janice Min Li, Kashif Ahmed, Kit Green-Sanderson, Mahnoor Rashid, Shahir M Morcos, Jayson T Maynes, Eric I Campos, Val C Sheffield, Ajoy Vincent, Elise Héon","doi":"10.1093/hmg/ddaf058","DOIUrl":null,"url":null,"abstract":"<p><p>A 29-year-old male presented with rod-cone degeneration leading to legal blindness, post-axial polydactyly, obesity, cognitive impairment, and fatty liver, features suggestive of a clinical diagnosis of Bardet-Biedl Syndrome (BBS). Following negative clinical genetic testing, genome analysis identified biallelic variants in IFT57: p.(Val397Glu) and p.(Lys225Asnfs*17). IFT57 is part of complex B of the intraflagellar transport (IFT) proteins, which is an adaptor to the anterograde transport of proteins, bringing cargo from the base of the primary cilia to the tip. Variants in IFT57 have not yet been associated with BBS or human retinal degeneration, but biallelic splicing variants were associated with a distinct ciliopathy: oral-facial-digital syndrome. Using patient-derived fibroblasts, IFT57-knockouts (KO) of RPE1, and mIMCD3 cells, we showed that p.(Lys225Asnfs*17) is subjected to non-sense mediated decay, and that p.(Val397Glu) is the predominant variant which leads to cilia defects. Exogenous expression of the p.(Val397Glu) variant partially restored structural and functional primary cilia defects, and of the anterograde transport in Ift57-KO mIMCD3 cells but it did not rescue primary cilia in retinal IFT57-KO-RPE1 cells. The cell autonomous effect, likely explains the retinal dystrophy in our proband with BBS.</p>","PeriodicalId":13070,"journal":{"name":"Human molecular genetics","volume":" ","pages":"1108-1122"},"PeriodicalIF":3.2000,"publicationDate":"2025-06-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12199350/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Human molecular genetics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1093/hmg/ddaf058","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
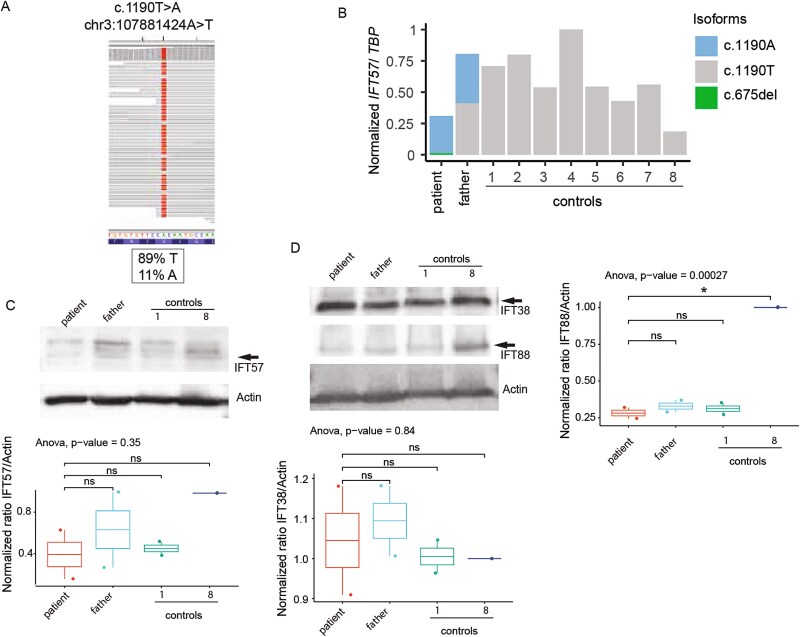
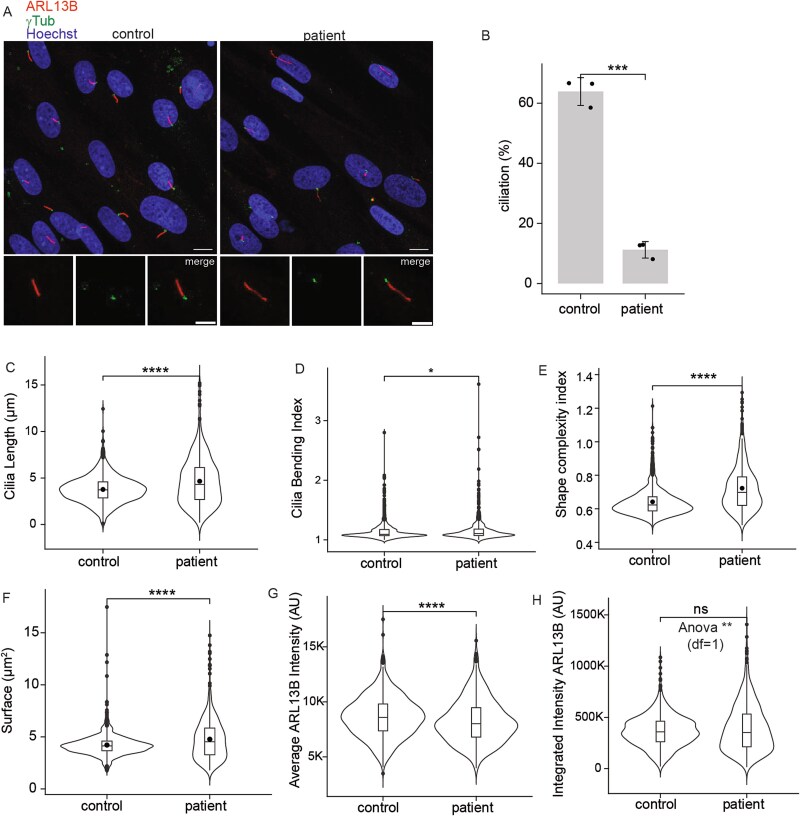
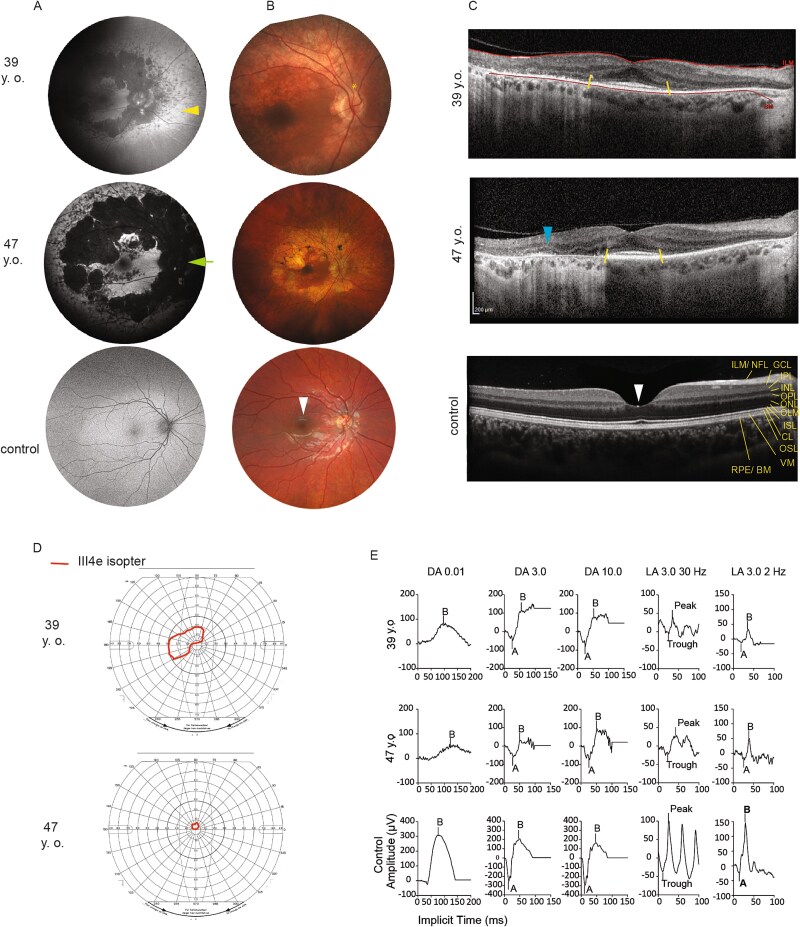
A 29-year-old male presented with rod-cone degeneration leading to legal blindness, post-axial polydactyly, obesity, cognitive impairment, and fatty liver, features suggestive of a clinical diagnosis of Bardet-Biedl Syndrome (BBS). Following negative clinical genetic testing, genome analysis identified biallelic variants in IFT57: p.(Val397Glu) and p.(Lys225Asnfs*17). IFT57 is part of complex B of the intraflagellar transport (IFT) proteins, which is an adaptor to the anterograde transport of proteins, bringing cargo from the base of the primary cilia to the tip. Variants in IFT57 have not yet been associated with BBS or human retinal degeneration, but biallelic splicing variants were associated with a distinct ciliopathy: oral-facial-digital syndrome. Using patient-derived fibroblasts, IFT57-knockouts (KO) of RPE1, and mIMCD3 cells, we showed that p.(Lys225Asnfs*17) is subjected to non-sense mediated decay, and that p.(Val397Glu) is the predominant variant which leads to cilia defects. Exogenous expression of the p.(Val397Glu) variant partially restored structural and functional primary cilia defects, and of the anterograde transport in Ift57-KO mIMCD3 cells but it did not rescue primary cilia in retinal IFT57-KO-RPE1 cells. The cell autonomous effect, likely explains the retinal dystrophy in our proband with BBS.
期刊介绍:
Human Molecular Genetics concentrates on full-length research papers covering a wide range of topics in all aspects of human molecular genetics. These include:
the molecular basis of human genetic disease
developmental genetics
cancer genetics
neurogenetics
chromosome and genome structure and function
therapy of genetic disease
stem cells in human genetic disease and therapy, including the application of iPS cells
genome-wide association studies
mouse and other models of human diseases
functional genomics
computational genomics
In addition, the journal also publishes research on other model systems for the analysis of genes, especially when there is an obvious relevance to human genetics.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


