Esther Uwibambe, Abdoulaye Yalcouyé, Elvis Twumasi Aboagye, Lettilia Xhakaza, Kalinka Popel, Norbert Dukuze, Thashi Bharadwaj, Carmen de Kock, Isabelle Schrauwen, Suzanne M Leal, Leon Mutesa, Ambroise Wonkam
{"title":"Exome sequencing revealed a novel homozygous variant in TRMT61 A in a multiplex family with atypical Cornelia de Lange Syndrome from Rwanda.","authors":"Esther Uwibambe, Abdoulaye Yalcouyé, Elvis Twumasi Aboagye, Lettilia Xhakaza, Kalinka Popel, Norbert Dukuze, Thashi Bharadwaj, Carmen de Kock, Isabelle Schrauwen, Suzanne M Leal, Leon Mutesa, Ambroise Wonkam","doi":"10.1186/s12920-025-02153-0","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>In 30% of patients who exhibit the clinical profile of Cornelia de Lange Syndrome (CdLS), the genetic cause remains undetermined. This proportion tends to be higher in low-resource settings including Africa. We performed a molecular characterization of CdLS in a multiplex Rwandan family.</p><p><strong>Methods: </strong>After a clinical evaluation of two affected siblings, DNA isolated from peripheral whole blood of the affected patients and their parents underwent Exome Sequencing (ES). Sanger sequencing validated the variant segregating with CdLS. In silico predictive tools, protein modelling, and cell-based experiments using HEK293T cells were used to investigate the pathogenicity of the variant found.</p><p><strong>Results: </strong>We identified a family with two parents and their two offspring (male and female), who were referred for hearing impairment. The 17-year-old female presented bilateral profound hearing impairment with moderate hypertelorism, progressive visual impairment, and secondary amenorrhea. The 14-year-old male displayed intellectual disability and a bilateral profound hearing impairment with no noticeable facial dysmorphism. Following exome sequencing (ES) of DNA samples obtained from the four family members, we found that the siblings harbored a novel likely pathogenic homozygous missense variant in the TRMT61 A gene [NM_152307.3:c.665C > T p.(Ala222Val)] inherited from both heterozygous parents. In silico analysis suggested that the variant substitutes a highly conserved amino acid, and 2-D structure modelling revealed a significant decrease in the stability of the protein. Cell-based experiment in HEK293T showed that the variant significantly affected the TRMT61 A protein localization which is thought to impact the mitochondrial and cytosolic functions.</p><p><strong>Conclusion: </strong>We reported a novel biallelic variant in TRMT61 A, [NM_152307.3:c.665C > T p.(Ala222Val)], which is associated with autosomal recessive atypical CdLS in a multiplex Rwandan family, the first report from Africa, and the second globally. The study emphasizes the need to expand the availability of ES for molecular characterization of rare diseases for the understudied genetically diverse population of Africa.</p>","PeriodicalId":8915,"journal":{"name":"BMC Medical Genomics","volume":"18 1","pages":"85"},"PeriodicalIF":2.0000,"publicationDate":"2025-05-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12070710/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Medical Genomics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s12920-025-02153-0","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
Background: In 30% of patients who exhibit the clinical profile of Cornelia de Lange Syndrome (CdLS), the genetic cause remains undetermined. This proportion tends to be higher in low-resource settings including Africa. We performed a molecular characterization of CdLS in a multiplex Rwandan family.
Methods: After a clinical evaluation of two affected siblings, DNA isolated from peripheral whole blood of the affected patients and their parents underwent Exome Sequencing (ES). Sanger sequencing validated the variant segregating with CdLS. In silico predictive tools, protein modelling, and cell-based experiments using HEK293T cells were used to investigate the pathogenicity of the variant found.
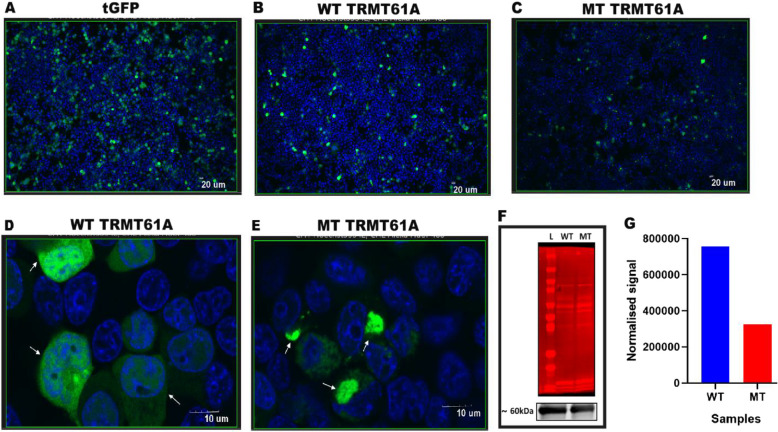
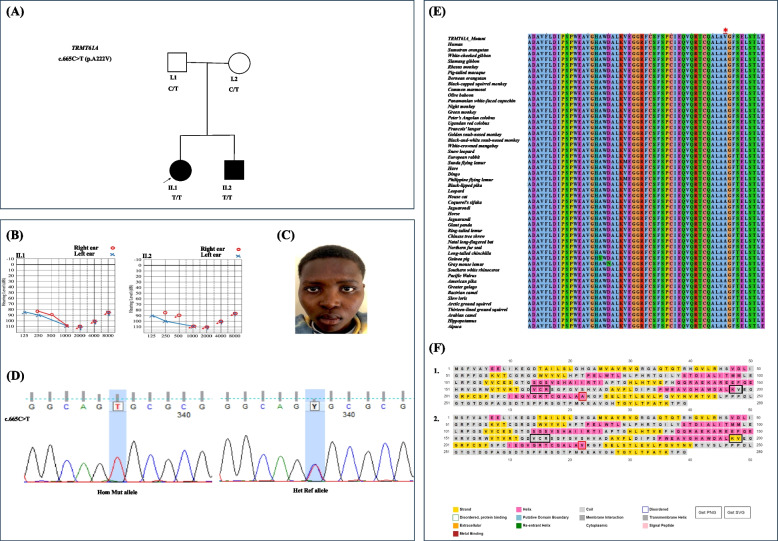
Results: We identified a family with two parents and their two offspring (male and female), who were referred for hearing impairment. The 17-year-old female presented bilateral profound hearing impairment with moderate hypertelorism, progressive visual impairment, and secondary amenorrhea. The 14-year-old male displayed intellectual disability and a bilateral profound hearing impairment with no noticeable facial dysmorphism. Following exome sequencing (ES) of DNA samples obtained from the four family members, we found that the siblings harbored a novel likely pathogenic homozygous missense variant in the TRMT61 A gene [NM_152307.3:c.665C > T p.(Ala222Val)] inherited from both heterozygous parents. In silico analysis suggested that the variant substitutes a highly conserved amino acid, and 2-D structure modelling revealed a significant decrease in the stability of the protein. Cell-based experiment in HEK293T showed that the variant significantly affected the TRMT61 A protein localization which is thought to impact the mitochondrial and cytosolic functions.
Conclusion: We reported a novel biallelic variant in TRMT61 A, [NM_152307.3:c.665C > T p.(Ala222Val)], which is associated with autosomal recessive atypical CdLS in a multiplex Rwandan family, the first report from Africa, and the second globally. The study emphasizes the need to expand the availability of ES for molecular characterization of rare diseases for the understudied genetically diverse population of Africa.
期刊介绍:
BMC Medical Genomics is an open access journal publishing original peer-reviewed research articles in all aspects of functional genomics, genome structure, genome-scale population genetics, epigenomics, proteomics, systems analysis, and pharmacogenomics in relation to human health and disease.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


