{"title":"What is the phenotype of heterozygous lipoprotein lipase deficiency?","authors":"Robert A Hegele","doi":"10.1097/MOL.0000000000000974","DOIUrl":null,"url":null,"abstract":"<p><strong>Purpose of review: </strong>Genetic testing of patients with severe hypertriglyceridemia often identifies a single heterozygous pathogenic variant in the LPL gene. The complex and variable phenotype associated with this genotype is the topic of this review.</p><p><strong>Recent findings: </strong>Previous research showed that heterozygosity for lipoprotein lipase deficiency is associated with reduced but variable post heparin lipolytic activity alongside inconsistent plasma lipid phenotypes ranging from normal to mild-to-moderate to severe hypertriglyceridemia. Recent research confirms and extends these observations, showing that a heterozygous individual can express a highly variable phenotype over time, depending on the presence of secondary factors. About 10% (range 8-20%) of patients with severe hypertriglyceridemia or multifactorial chylomicronemia syndrome are heterozygous for a rare pathogenic LPL variant, and a clinically relevant minority of these has recalcitrant or sustained hypertriglyceridemia.</p><p><strong>Summary: </strong>Heterozygosity for lipoprotein lipase deficiency predisposes to hypertriglyceridemia, which is sometimes severe depending on secondary factors, but is typically quite responsive to routine interventions such as diet, lifestyle and existing lipid-lowering therapies. However, many heterozygotes for pathogenic variants in LPL have completely normal plasma lipids.</p>","PeriodicalId":11109,"journal":{"name":"Current opinion in lipidology","volume":"36 2","pages":"96-103"},"PeriodicalIF":4.6000,"publicationDate":"2025-04-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11888829/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Current opinion in lipidology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1097/MOL.0000000000000974","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/15 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Purpose of review: Genetic testing of patients with severe hypertriglyceridemia often identifies a single heterozygous pathogenic variant in the LPL gene. The complex and variable phenotype associated with this genotype is the topic of this review.
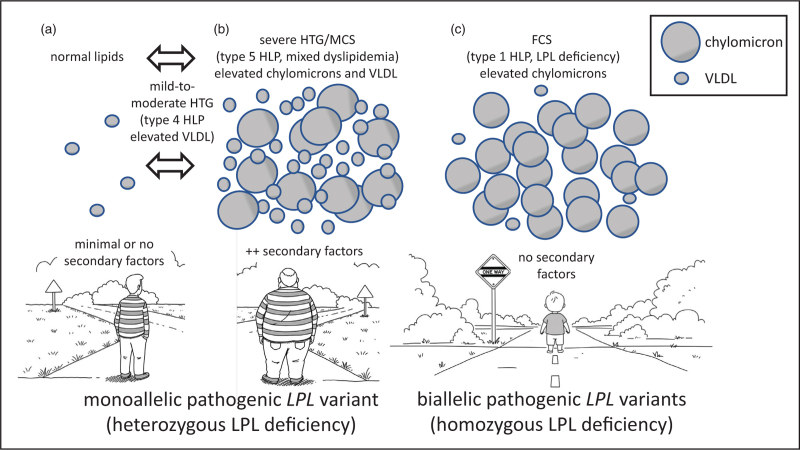
Recent findings: Previous research showed that heterozygosity for lipoprotein lipase deficiency is associated with reduced but variable post heparin lipolytic activity alongside inconsistent plasma lipid phenotypes ranging from normal to mild-to-moderate to severe hypertriglyceridemia. Recent research confirms and extends these observations, showing that a heterozygous individual can express a highly variable phenotype over time, depending on the presence of secondary factors. About 10% (range 8-20%) of patients with severe hypertriglyceridemia or multifactorial chylomicronemia syndrome are heterozygous for a rare pathogenic LPL variant, and a clinically relevant minority of these has recalcitrant or sustained hypertriglyceridemia.
Summary: Heterozygosity for lipoprotein lipase deficiency predisposes to hypertriglyceridemia, which is sometimes severe depending on secondary factors, but is typically quite responsive to routine interventions such as diet, lifestyle and existing lipid-lowering therapies. However, many heterozygotes for pathogenic variants in LPL have completely normal plasma lipids.
期刊介绍:
With its easy-to-digest reviews on important advances in world literature, Current Opinion in Lipidology offers expert evaluation on a wide range of topics from six key disciplines including nutrition and metabolism, genetics and molecular biology, and hyperlipidaemia and cardiovascular disease. Published bimonthly, each issue covers in detail the most pertinent advances in these fields from the previous year. This is supplemented by a section of Bimonthly Updates, which deliver an insight into new developments at the cutting edge of the disciplines covered in the journal.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


