Functional analysis of a novel FOXL2 mutation in blepharophimosis, ptosis, and epicanthus inversus syndrome type II and elucidation of the genotype-phenotype correlation.
Bingyan Shen, Xi Chen, Xiuying Zhu, Ziwen Chen, Yenan Fang, Qin Dai, Xinyu Li, Qiqi Xie, Wencan Wu, Min Wang
{"title":"Functional analysis of a novel FOXL2 mutation in blepharophimosis, ptosis, and epicanthus inversus syndrome type II and elucidation of the genotype-phenotype correlation.","authors":"Bingyan Shen, Xi Chen, Xiuying Zhu, Ziwen Chen, Yenan Fang, Qin Dai, Xinyu Li, Qiqi Xie, Wencan Wu, Min Wang","doi":"10.1186/s40246-025-00752-7","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Blepharophimosis, ptosis, and epicanthus inversus syndrome (BPES) is a rare autosomal dominant disorder caused by genetic mutations. However, the genotype-phenotype correlation remains unclear. This study aimed to identify mutations in a Chinese family with BPES and elucidate the genotype-phenotype relationship.</p><p><strong>Methods: </strong>A comprehensive clinical and molecular genetic analysis was conducted on a three-generation Chinese family with BPES, which was prospectively enrolled at the Eye Hospital of Wenzhou Medical University. Affected individuals underwent systematic phenotyping, including detailed physical and ophthalmic evaluations. Genomic DNA was isolated from peripheral blood samples and subjected to whole-exome sequencing, followed by targeted Sanger sequencing for variant validation. Candidate disease-associated variants were analyzed using in silico predictive algorithms to assess their potential structural and functional impact on encoded proteins. To further elucidate the pathogenicity of the identified mutation, functional studies were performed, including immunofluorescence-based subcellular localization assays and quantitative real-time PCR to evaluate transcriptional regulatory effects.</p><p><strong>Results: </strong>Six affected individuals of this pedigree presented with canonical BPES features including small palpebral fissures, ptosis, epicanthus inversus, and telecanthus, without premature ovarian failure, consistent with a diagnosis of BPES type II. Whole-exome sequencing revealed a heterozygous missense mutation (c.313 A > C:p.N105H) in FOXL2, which was subsequently validated by Sanger sequencing. This variant demonstrated complete cosegregation with the BPES phenotype across all affected family members. According to ACMG guidelines, the variant was classified as Likely Pathogenic (PS1 + PM1 + PM2 + PP3). In silico pathogenicity prediction tools classified the p.N105H variant as deleterious. Immunofluorescence assays revealed aberrant nuclear aggregation of the mutant FOXL2 protein, and functional characterization via quantitative real-time PCR demonstrated no significant dysregulation (P > 0.05) of downstream targets (STAR, OSR2).</p><p><strong>Conclusions: </strong>This study provides functional evidence of the pathogenic FOXL2 mutation (c.313 A > C, p.N105H) in BPES type II, demonstrating its disruptive effects on protein localization while maintaining normal transcriptional activity of downstream targets. These findings expand the mutational spectrum of FOXL2 related disorders and enhance our understanding of genotype-phenotype correlations in BPES.</p>","PeriodicalId":13183,"journal":{"name":"Human Genomics","volume":"19 1","pages":"41"},"PeriodicalIF":4.3000,"publicationDate":"2025-04-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12008864/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Human Genomics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s40246-025-00752-7","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Blepharophimosis, ptosis, and epicanthus inversus syndrome (BPES) is a rare autosomal dominant disorder caused by genetic mutations. However, the genotype-phenotype correlation remains unclear. This study aimed to identify mutations in a Chinese family with BPES and elucidate the genotype-phenotype relationship.
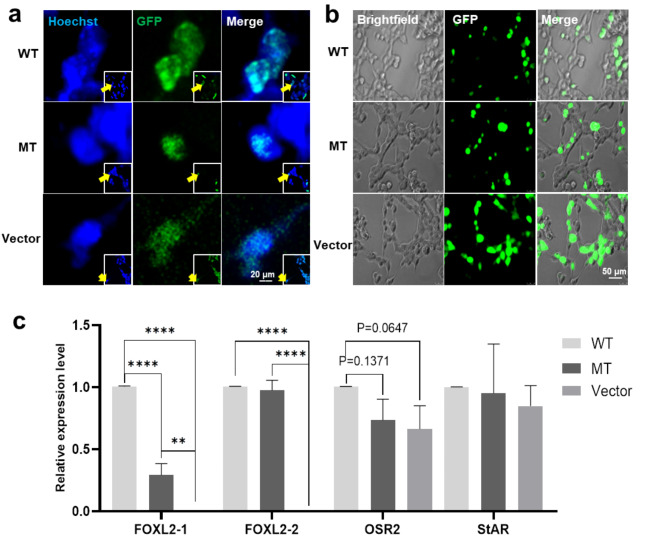
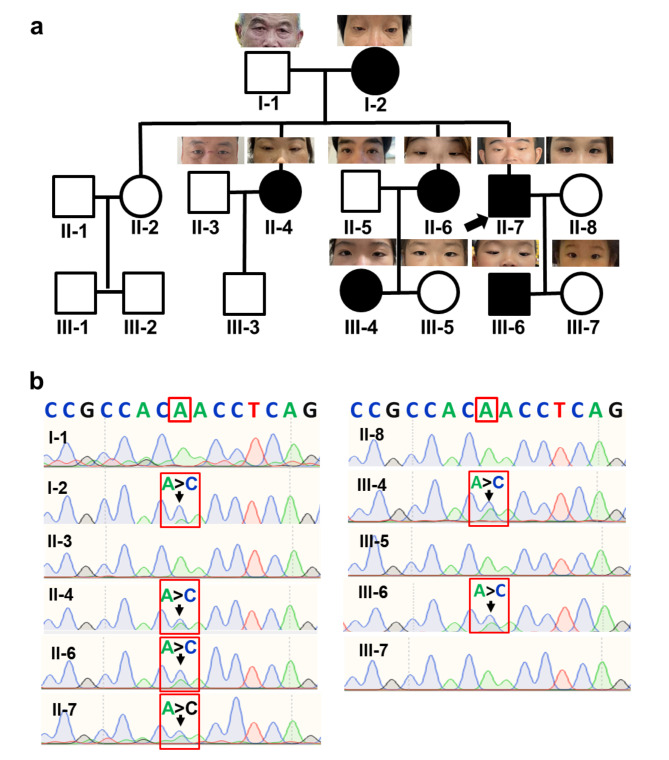
Methods: A comprehensive clinical and molecular genetic analysis was conducted on a three-generation Chinese family with BPES, which was prospectively enrolled at the Eye Hospital of Wenzhou Medical University. Affected individuals underwent systematic phenotyping, including detailed physical and ophthalmic evaluations. Genomic DNA was isolated from peripheral blood samples and subjected to whole-exome sequencing, followed by targeted Sanger sequencing for variant validation. Candidate disease-associated variants were analyzed using in silico predictive algorithms to assess their potential structural and functional impact on encoded proteins. To further elucidate the pathogenicity of the identified mutation, functional studies were performed, including immunofluorescence-based subcellular localization assays and quantitative real-time PCR to evaluate transcriptional regulatory effects.
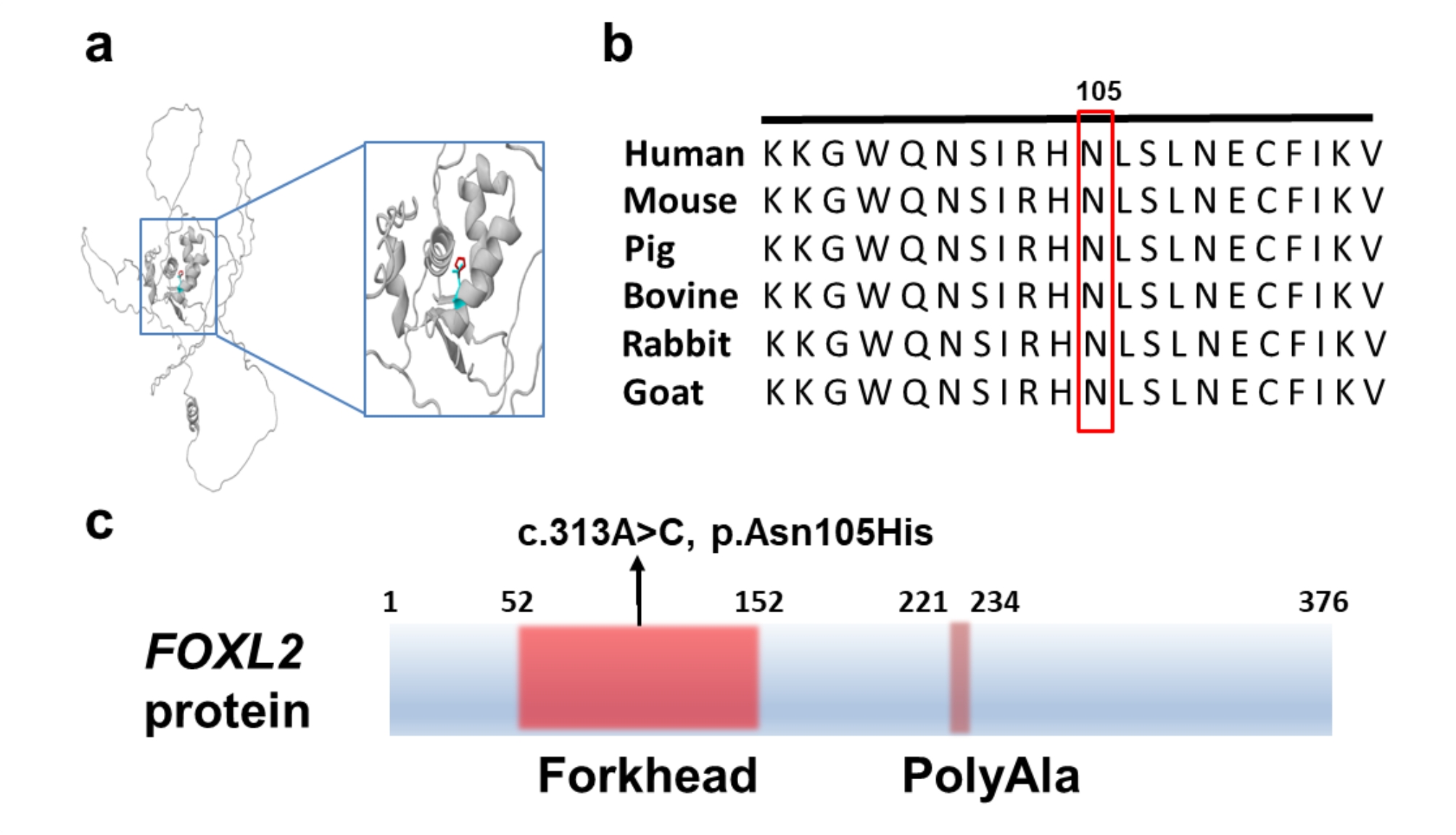
Results: Six affected individuals of this pedigree presented with canonical BPES features including small palpebral fissures, ptosis, epicanthus inversus, and telecanthus, without premature ovarian failure, consistent with a diagnosis of BPES type II. Whole-exome sequencing revealed a heterozygous missense mutation (c.313 A > C:p.N105H) in FOXL2, which was subsequently validated by Sanger sequencing. This variant demonstrated complete cosegregation with the BPES phenotype across all affected family members. According to ACMG guidelines, the variant was classified as Likely Pathogenic (PS1 + PM1 + PM2 + PP3). In silico pathogenicity prediction tools classified the p.N105H variant as deleterious. Immunofluorescence assays revealed aberrant nuclear aggregation of the mutant FOXL2 protein, and functional characterization via quantitative real-time PCR demonstrated no significant dysregulation (P > 0.05) of downstream targets (STAR, OSR2).
Conclusions: This study provides functional evidence of the pathogenic FOXL2 mutation (c.313 A > C, p.N105H) in BPES type II, demonstrating its disruptive effects on protein localization while maintaining normal transcriptional activity of downstream targets. These findings expand the mutational spectrum of FOXL2 related disorders and enhance our understanding of genotype-phenotype correlations in BPES.
期刊介绍:
Human Genomics is a peer-reviewed, open access, online journal that focuses on the application of genomic analysis in all aspects of human health and disease, as well as genomic analysis of drug efficacy and safety, and comparative genomics.
Topics covered by the journal include, but are not limited to: pharmacogenomics, genome-wide association studies, genome-wide sequencing, exome sequencing, next-generation deep-sequencing, functional genomics, epigenomics, translational genomics, expression profiling, proteomics, bioinformatics, animal models, statistical genetics, genetic epidemiology, human population genetics and comparative genomics.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


