The pathogenesis, clinical presentations and treatment of monogenic systemic vasculitis
IF 32.7
1区 医学
Q1 RHEUMATOLOGY
引用次数: 0
Abstract
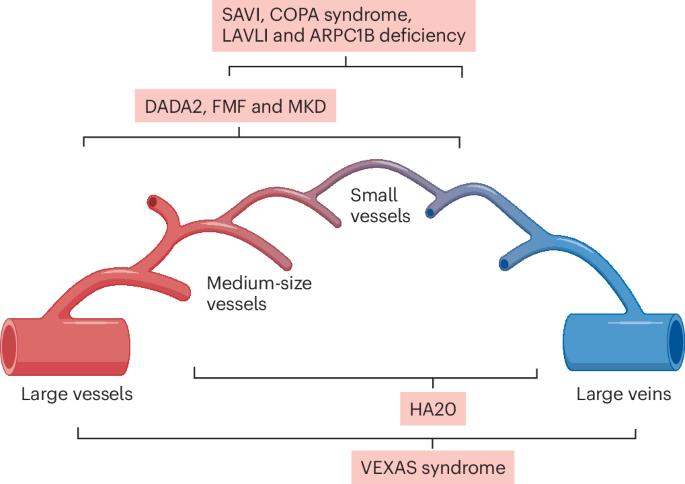
Many monogenic autoinflammatory diseases, including DADA2 (deficiency of adenosine deaminase 2), HA20 (haploinsufficiency of A20), SAVI (STING-associated vasculopathy with onset in infancy), COPA syndrome, LAVLI (LYN kinase-associated vasculopathy and liver fibrosis) and VEXAS (vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic) syndrome, present predominantly with vasculitis and constitute a substantial subgroup of vasculitic conditions associated with a ‘probable aetiology’. The spectrum of monogenic vasculitis encompasses all sizes and types of blood vessel, ranging from large vessels to medium-size and small vessels, and from the arterial side to the venous side of the vasculature. Monogenic vasculitis typically starts early in life during infancy or childhood; VEXAS syndrome, which presents in late adulthood, is an exception. The activation of myeloid cells via inflammasome and nuclear factor-κB pathways, type I interferon-enhanced autoimmune mechanisms and/or dysregulated adaptive immune responses have an important role in the development of immune-mediated endothelial dysfunction and vascular damage. Genetic testing is essential for the diagnosis of underlying monogenic autoinflammatory diseases; however, the penetrance of genetic variants can vary. Increased awareness and recognition of distinctive clinical findings could facilitate earlier diagnosis and allow for more-targeted treatments. This Review discusses the clinical features, pathogenesis, diagnosis and management of monogenic forms of vasculitis. The authors emphasize that increased awareness of these rare diseases could aid earlier diagnosis and better, more-targeted treatment options for patients.

单基因系统性血管炎的发病机制、临床表现及治疗
许多单基因自身炎症性疾病,包括DADA2(腺苷脱氨酶2缺乏症)、HA20 (A20单倍功能不全)、SAVI(婴儿期发作的sting相关血管病变)、COPA综合征、LAVLI (LYN激酶相关血管病变和肝纤维化)和VEXAS(空泡、E1酶、x连锁、自身炎症、体)综合征,主要表现为血管炎,构成了与“可能病因”相关的血管疾病的一个重要亚群。单基因性血管炎包括所有大小和类型的血管,从大血管到中、小血管,从动脉侧到静脉侧。单基因血管炎通常在婴儿期或儿童期早期开始;在成年后期出现的VEXAS综合征是一个例外。髓细胞通过炎性体和核因子-κB途径激活、I型干扰素增强的自身免疫机制和/或失调的适应性免疫反应在免疫介导的内皮功能障碍和血管损伤的发展中起重要作用。基因检测对于诊断潜在的单基因自身炎症性疾病至关重要;然而,基因变异的外显率是不同的。提高对独特临床表现的认识和认识可以促进早期诊断,并允许更有针对性的治疗。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Nature Reviews Rheumatology
医学-风湿病学
CiteScore
29.90
自引率
0.90%
发文量
137
审稿时长
6-12 weeks
期刊介绍:
Nature Reviews Rheumatology is part of the Nature Reviews portfolio of journals. The journal scope covers the entire spectrum of rheumatology research. We ensure that our articles are accessible to the widest possible audience.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


