{"title":"Phylodynamics of hepatitis B virus genotype B in East Asia: A population genomics analysis.","authors":"Serena Y C Lin, Patrick C Y Woo","doi":"10.14440/jbm.2025.0084","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Hepatitis B virus (HBV) genotype B (HBV/B) is the predominant strain in Taiwan and several East Asian countries.</p><p><strong>Objective: </strong>The aim of this study is to use comprehensive phylogenetic analysis tools to monitor the long-term molecular evolution dynamic of HBV genotype B population in East Asia.</p><p><strong>Methods: </strong>In this study, full genome sequences of HBV with temporal information were extracted from GenBank and analyzed using the Bayesian Markov chain Monte Carlo method to identify best-fitting coalescent models.</p><p><strong>Results: </strong>Bayesian Skygrid analysis revealed a viral effective population (phylodynamic) bottleneck for HBV/B in 2003, a pattern similar to the previously described HBV genotype C (HBV/C). Despite these similarities, the viral dynamics for HBV/B and HBV/C diverged after 2005. HBV/C exhibited a marked decrease in genetic diversity across East Asia, whereas HBV/B maintained stable genetic diversity after 2005. Phylogeographic analysis using Neighbor-Joining and Bayesian maximum clade credibility trees indicated that Taiwan was likely the geographic origin of the most recent common ancestor of HBV/B in East Asia. An early clade spread to Japan and subsequently to the West Coast of the United States of America. Another clade dispersed to China, spread widely across the region, and was reintroduced to Taiwan multiple times. In contrast, HBV/C likely originated in China and spread to Japan, Korea, and Taiwan over several decades.</p><p><strong>Conclusion: </strong>This study highlights the similarities and differences between the viral dynamics and geographical evolutionary pathways between HBV/B and HBV/C.</p>","PeriodicalId":73618,"journal":{"name":"Journal of biological methods","volume":"12 1","pages":"e99010048"},"PeriodicalIF":0.0000,"publicationDate":"2025-01-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11973052/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of biological methods","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.14440/jbm.2025.0084","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Hepatitis B virus (HBV) genotype B (HBV/B) is the predominant strain in Taiwan and several East Asian countries.
Objective: The aim of this study is to use comprehensive phylogenetic analysis tools to monitor the long-term molecular evolution dynamic of HBV genotype B population in East Asia.
Methods: In this study, full genome sequences of HBV with temporal information were extracted from GenBank and analyzed using the Bayesian Markov chain Monte Carlo method to identify best-fitting coalescent models.
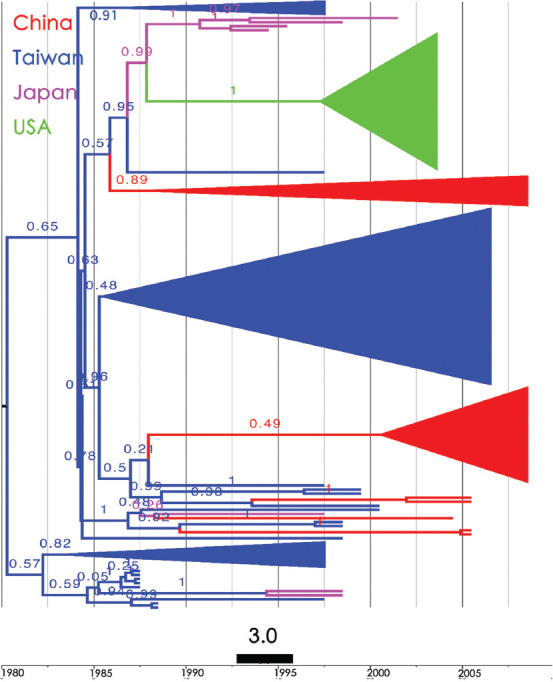
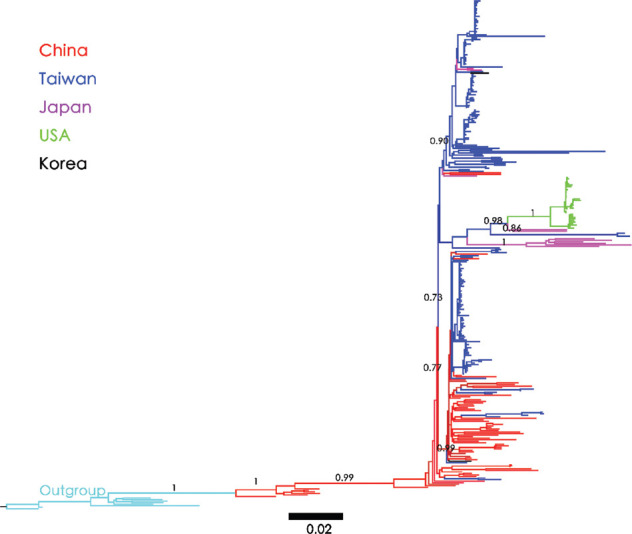
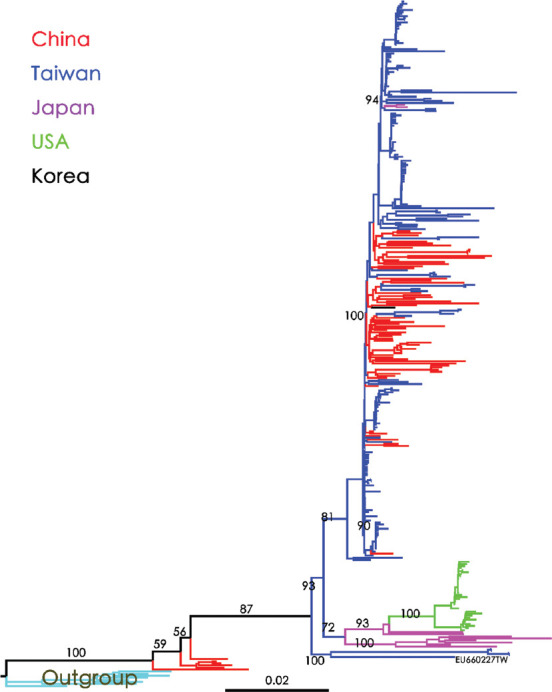
Results: Bayesian Skygrid analysis revealed a viral effective population (phylodynamic) bottleneck for HBV/B in 2003, a pattern similar to the previously described HBV genotype C (HBV/C). Despite these similarities, the viral dynamics for HBV/B and HBV/C diverged after 2005. HBV/C exhibited a marked decrease in genetic diversity across East Asia, whereas HBV/B maintained stable genetic diversity after 2005. Phylogeographic analysis using Neighbor-Joining and Bayesian maximum clade credibility trees indicated that Taiwan was likely the geographic origin of the most recent common ancestor of HBV/B in East Asia. An early clade spread to Japan and subsequently to the West Coast of the United States of America. Another clade dispersed to China, spread widely across the region, and was reintroduced to Taiwan multiple times. In contrast, HBV/C likely originated in China and spread to Japan, Korea, and Taiwan over several decades.
Conclusion: This study highlights the similarities and differences between the viral dynamics and geographical evolutionary pathways between HBV/B and HBV/C.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


