Haonan Xie, Biao Chen, Chunnian He, Chunsheng Shi, Enzuo Liu and Naiqin Zhao
{"title":"Unveiling the reaction selectivity mechanism of molybdenum and tungsten carbides as cathode catalysts for Li–CO2 batteries†","authors":"Haonan Xie, Biao Chen, Chunnian He, Chunsheng Shi, Enzuo Liu and Naiqin Zhao","doi":"10.1039/D5TA00211G","DOIUrl":null,"url":null,"abstract":"<p >The type of cathode catalysts and the adsorption behavior of molecules on the surface play a decisive role in the selectivity of the lithium–CO<small><sub>2</sub></small> battery electrochemical reaction. However, few researchers have revealed the regulatory mechanism from the perspective of electronic structures. In this work, the paths and products of the Li–CO<small><sub>2</sub></small> electrochemical reaction on Mo<small><sub>2</sub></small>C(101) and W<small><sub>2</sub></small>C(101) have been investigated by using the first-principles calculation. A surface covered by a molecular layer of CO<small><sub>2</sub></small> is proposed, and the feasibility of the model is thermodynamically proved. Based on the covered surface model, the reaction selectivity is consistent with the experimental results. Combined with electronic structure analysis, it is revealed that the d-orbital electrons of Mo in Mo<small><sub>2</sub></small>C(101) are further activated after binding to CO<small><sub>2</sub></small>. Metastable oxalate stabilization is achieved by enhancing the contribution of ionic bond components in the chemical bond with the key intermediate oxalate. The delocalized W-d orbital in W<small><sub>2</sub></small>C(101) is hybridized with the p orbitals of C and O in *CO<small><sub>2</sub></small>, weakening the C<img>O bond in CO<small><sub>2</sub></small> and promoting the formation of carbonate. It provides a new idea for the construction of a Li–CO<small><sub>2</sub></small> battery reaction model and understanding the battery reaction selectivity.</p>","PeriodicalId":82,"journal":{"name":"Journal of Materials Chemistry A","volume":" 19","pages":" 13898-13908"},"PeriodicalIF":9.5000,"publicationDate":"2025-04-07","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Materials Chemistry A","FirstCategoryId":"88","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2025/ta/d5ta00211g","RegionNum":2,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
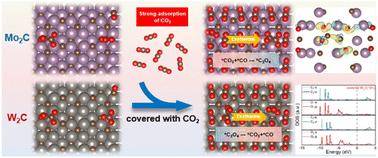
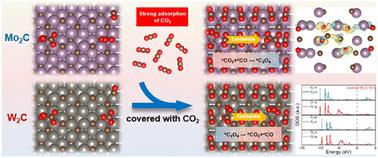
The type of cathode catalysts and the adsorption behavior of molecules on the surface play a decisive role in the selectivity of the lithium–CO2 battery electrochemical reaction. However, few researchers have revealed the regulatory mechanism from the perspective of electronic structures. In this work, the paths and products of the Li–CO2 electrochemical reaction on Mo2C(101) and W2C(101) have been investigated by using the first-principles calculation. A surface covered by a molecular layer of CO2 is proposed, and the feasibility of the model is thermodynamically proved. Based on the covered surface model, the reaction selectivity is consistent with the experimental results. Combined with electronic structure analysis, it is revealed that the d-orbital electrons of Mo in Mo2C(101) are further activated after binding to CO2. Metastable oxalate stabilization is achieved by enhancing the contribution of ionic bond components in the chemical bond with the key intermediate oxalate. The delocalized W-d orbital in W2C(101) is hybridized with the p orbitals of C and O in *CO2, weakening the CO bond in CO2 and promoting the formation of carbonate. It provides a new idea for the construction of a Li–CO2 battery reaction model and understanding the battery reaction selectivity.
期刊介绍:
The Journal of Materials Chemistry A, B & C covers a wide range of high-quality studies in the field of materials chemistry, with each section focusing on specific applications of the materials studied. Journal of Materials Chemistry A emphasizes applications in energy and sustainability, including topics such as artificial photosynthesis, batteries, and fuel cells. Journal of Materials Chemistry B focuses on applications in biology and medicine, while Journal of Materials Chemistry C covers applications in optical, magnetic, and electronic devices. Example topic areas within the scope of Journal of Materials Chemistry A include catalysis, green/sustainable materials, sensors, and water treatment, among others.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


