Severe Joubert syndrome in family with homozygous POC1B p.Arg106Pro variant is due to a co-inherited deep-intronic mutation in the neighboring CEP290 gene.
Christian Betz, Björn Reusch, Thomas Langmann, Sandra Habbig, Bodo B Beck, Hanno J Bolz
{"title":"Severe Joubert syndrome in family with homozygous POC1B p.Arg106Pro variant is due to a co-inherited deep-intronic mutation in the neighboring CEP290 gene.","authors":"Christian Betz, Björn Reusch, Thomas Langmann, Sandra Habbig, Bodo B Beck, Hanno J Bolz","doi":"10.1016/j.xhgg.2025.100429","DOIUrl":null,"url":null,"abstract":"<p><p>\"En bloc\" inheritance of point mutations in adjacent genes has rarely been described. We have previously reported a family with severe, mostly early-lethal Joubert syndrome (JBTS) with early-onset severe retinal dystrophy (EOSRD) and polycystic kidney disease (PKD), which at that time had been attributed to a homozygous pathogenic missense variant, p.Arg106Pro (c.317G>C), in the ciliary POC1B gene. Because this and other POC1B variants were, in subsequent studies, only reported in patients with non-syndromic childhood or early-adult-onset macular dystrophy, we have now reassessed our index patient by long-read high-fidelity (HiFi) whole-genome sequencing (LR-WGS). We identified a homozygous deep-intronic variant, c.2818-657T>G, in CEP290, a JBTS/Meckel syndrome-associated gene on chromosome 12q21, only 1.28 Mb from the N terminus of POC1B. cDNA analysis revealed aberrant splicing with the frame-shifting inclusion of 37 bp from CEP290 intron 25, predicting the loss of CEP290 function. EOSRD and PKD can fully be ascribed to this CEP290 variant, whose effect outshines the \"background\" non-syndromic POC1B retinopathy and co-segregates with the severe syndromic phenotype. Our novel findings in this family no longer justify POC1B as a JBTS gene. This co-inheritance of two ciliopathies, with the clinically decisive variant hidden deep in an intron, exemplifies the importance of WGS for achieving the complete diagnosis in challenging cases.</p>","PeriodicalId":34530,"journal":{"name":"HGG Advances","volume":" ","pages":"100429"},"PeriodicalIF":3.6000,"publicationDate":"2025-07-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12018183/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"HGG Advances","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1016/j.xhgg.2025.100429","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/3/31 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
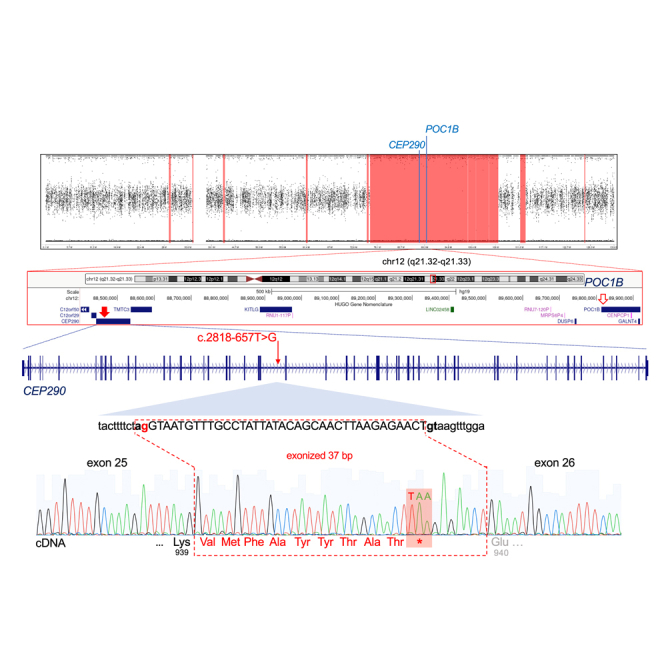
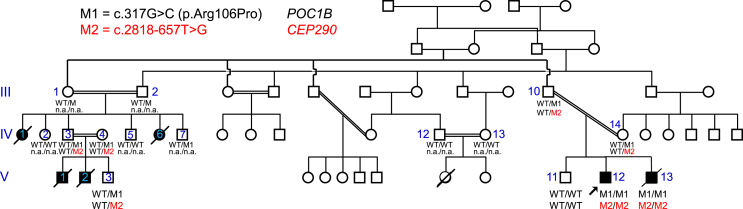
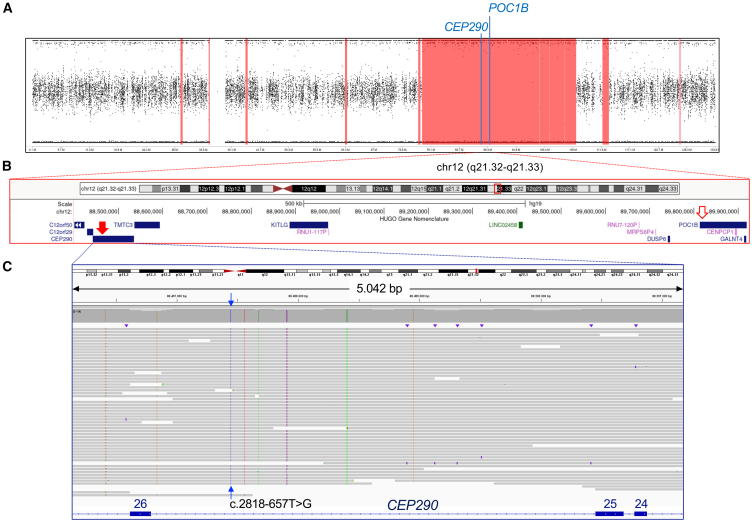
"En bloc" inheritance of point mutations in adjacent genes has rarely been described. We have previously reported a family with severe, mostly early-lethal Joubert syndrome (JBTS) with early-onset severe retinal dystrophy (EOSRD) and polycystic kidney disease (PKD), which at that time had been attributed to a homozygous pathogenic missense variant, p.Arg106Pro (c.317G>C), in the ciliary POC1B gene. Because this and other POC1B variants were, in subsequent studies, only reported in patients with non-syndromic childhood or early-adult-onset macular dystrophy, we have now reassessed our index patient by long-read high-fidelity (HiFi) whole-genome sequencing (LR-WGS). We identified a homozygous deep-intronic variant, c.2818-657T>G, in CEP290, a JBTS/Meckel syndrome-associated gene on chromosome 12q21, only 1.28 Mb from the N terminus of POC1B. cDNA analysis revealed aberrant splicing with the frame-shifting inclusion of 37 bp from CEP290 intron 25, predicting the loss of CEP290 function. EOSRD and PKD can fully be ascribed to this CEP290 variant, whose effect outshines the "background" non-syndromic POC1B retinopathy and co-segregates with the severe syndromic phenotype. Our novel findings in this family no longer justify POC1B as a JBTS gene. This co-inheritance of two ciliopathies, with the clinically decisive variant hidden deep in an intron, exemplifies the importance of WGS for achieving the complete diagnosis in challenging cases.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


