{"title":"Genetic and Clinical Characterization of Complex Glycerol Kinase Deficiency in Two Male Siblings: A Case Report.","authors":"Kakha Bregvadze, Nino Kheladze, Nana Nino Tatishvili, Nino Dikhaminjia, Mariam Ghughunishvili, Shorena Tchankvetadze, Tinatin Tkemaladze","doi":"10.1177/11795514251317419","DOIUrl":null,"url":null,"abstract":"<p><p>Complex glycerol kinase deficiency (CGKD), also known as Xp21 contiguous gene deletion syndrome, is a rare X-linked recessive disorder resulting from partial deletion of the Xp21.3 chromosomal region. CGKD encompasses several loci, including glycerol kinase (<i>GK</i>), Duchenne muscular dystrophy (<i>DMD</i>), X-linked adrenal hypoplasia congenita (<i>NR0B1</i>), and intellectual developmental disorder (<i>IL1RAPL1</i>). We present the cases of two male siblings diagnosed with CGKD. The elder sibling was initially suspected of having congenital adrenal hypoplasia (CAH). Whole exome sequencing (WES) revealed an interstitial deletion of 6.6 Mb on Xp21.3p21.1, encompassing critical genes including <i>GK</i>, <i>DMD</i>, <i>NR0B1</i>, and <i>IL1RAPL1.</i> The younger sibling was diagnosed shortly after birth based on family history, clinical and biochemical findings. The presented report highlights the diagnostic challenges associated with CGKD and the important role of genetic testing in confirming the diagnosis. A multidisciplinary team approach is necessary.</p>","PeriodicalId":44715,"journal":{"name":"Clinical Medicine Insights-Endocrinology and Diabetes","volume":"18 ","pages":"11795514251317419"},"PeriodicalIF":3.0000,"publicationDate":"2025-03-31","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11960146/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Clinical Medicine Insights-Endocrinology and Diabetes","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1177/11795514251317419","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"Q3","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
引用次数: 0
Abstract
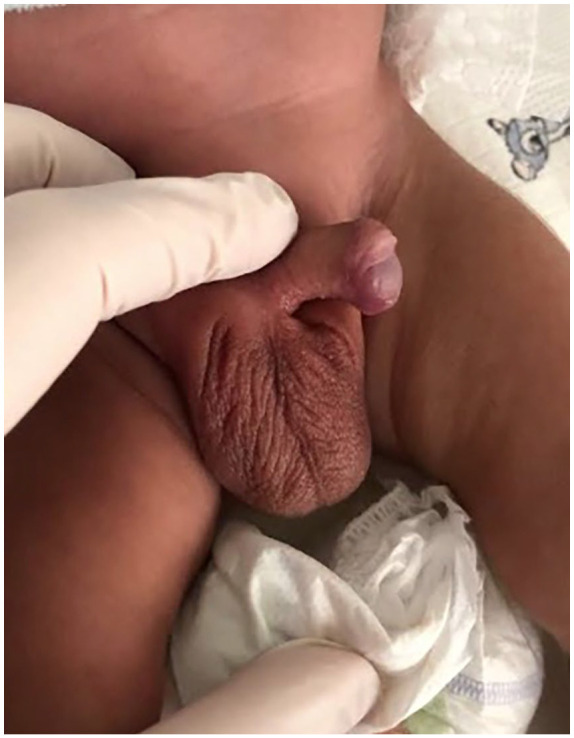
Complex glycerol kinase deficiency (CGKD), also known as Xp21 contiguous gene deletion syndrome, is a rare X-linked recessive disorder resulting from partial deletion of the Xp21.3 chromosomal region. CGKD encompasses several loci, including glycerol kinase (GK), Duchenne muscular dystrophy (DMD), X-linked adrenal hypoplasia congenita (NR0B1), and intellectual developmental disorder (IL1RAPL1). We present the cases of two male siblings diagnosed with CGKD. The elder sibling was initially suspected of having congenital adrenal hypoplasia (CAH). Whole exome sequencing (WES) revealed an interstitial deletion of 6.6 Mb on Xp21.3p21.1, encompassing critical genes including GK, DMD, NR0B1, and IL1RAPL1. The younger sibling was diagnosed shortly after birth based on family history, clinical and biochemical findings. The presented report highlights the diagnostic challenges associated with CGKD and the important role of genetic testing in confirming the diagnosis. A multidisciplinary team approach is necessary.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


