{"title":"From benign neurofibromas to malignant peripheral nerve sheath tumors (MPNST): a gaming among multiple factors.","authors":"Yanan Yu, Chengjiang Wei, Minghui Yue, Cheng Zhang, Yixiao Wang, Zhichao Wang","doi":"10.1007/s13402-025-01054-9","DOIUrl":null,"url":null,"abstract":"<p><p>Almost all patients of Neurofibromatosis Type I (NF1) develop benign peripheral nerve tumors called neurofibromas, which are derived from neural crest Schwann cell lineage progenitors with biallelic NF1 gene mutations. More than 90% of NF1 patients develop dermal neurofibromas (DN), and 25-50% develop plexiform neurofibromas (PN). In 8-13% of individuals with NF1, PN can transform into malignant peripheral nerve sheath tumors (MPNSTs), a type of nerve soft tissue sarcoma that is the main cause of mortality of NF1 patients. In addition to arising from benign neurofibromas (50%), MPNSTs can also occur spontaneously (~40%) or following radiation therapy (~10%). Treatment for MPNST is limited to complete resection with negative margins. Still, the high recurrence of MPNST is a major concern. However, full resection of the pre-malignant lesions can largely reduce the recurrence and mortality of patients. So, early diagnosis and distinguishing malignancy from benign and premalignant lesions are particularly important. During the progression from benign neurofibromas to malignancy, a variety of changes including tumor morphology, genetic mutations, expression of multiple signaling pathways-related proteins and genome instability gradually occur. In this review, we detail these changes with the goals of identifying the histological and/or molecular signs of malignancy initiation, and an optimal therapeutic intervention window, to inhibit tumor progression and reduce the rate of mortality.</p>","PeriodicalId":9690,"journal":{"name":"Cellular Oncology","volume":" ","pages":"841-857"},"PeriodicalIF":4.8000,"publicationDate":"2025-08-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12238183/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cellular Oncology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s13402-025-01054-9","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/4/2 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"Medicine","Score":null,"Total":0}
引用次数: 0
Abstract
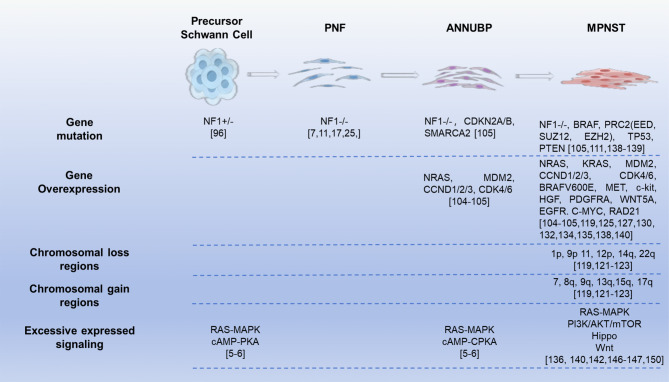
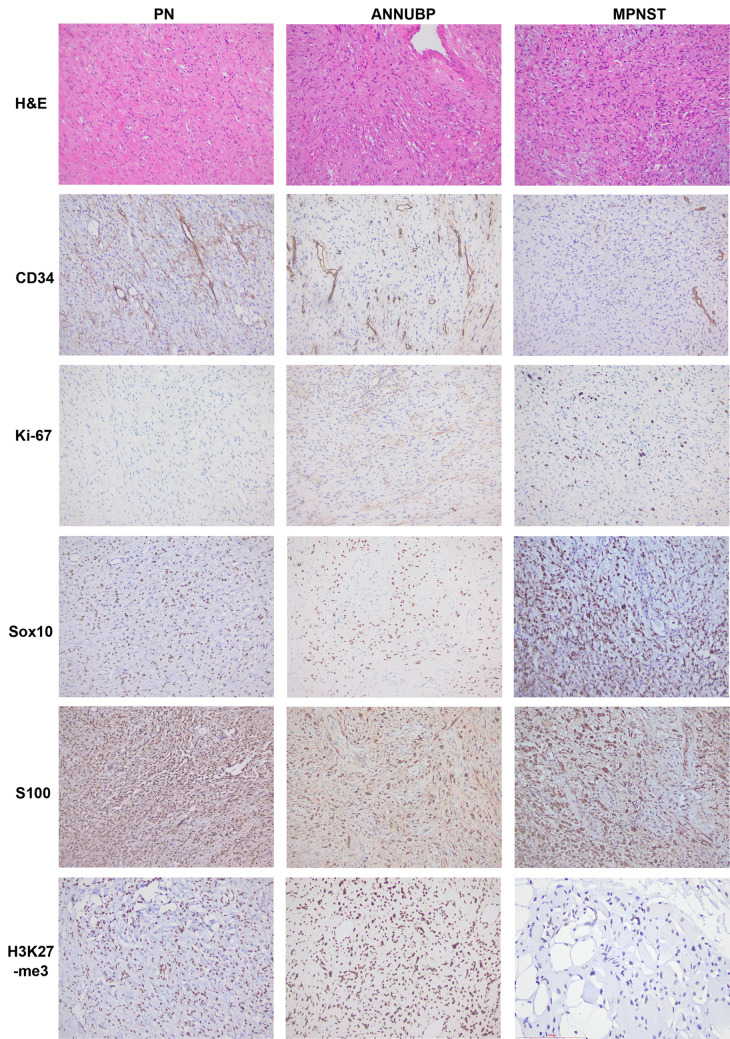
Almost all patients of Neurofibromatosis Type I (NF1) develop benign peripheral nerve tumors called neurofibromas, which are derived from neural crest Schwann cell lineage progenitors with biallelic NF1 gene mutations. More than 90% of NF1 patients develop dermal neurofibromas (DN), and 25-50% develop plexiform neurofibromas (PN). In 8-13% of individuals with NF1, PN can transform into malignant peripheral nerve sheath tumors (MPNSTs), a type of nerve soft tissue sarcoma that is the main cause of mortality of NF1 patients. In addition to arising from benign neurofibromas (50%), MPNSTs can also occur spontaneously (~40%) or following radiation therapy (~10%). Treatment for MPNST is limited to complete resection with negative margins. Still, the high recurrence of MPNST is a major concern. However, full resection of the pre-malignant lesions can largely reduce the recurrence and mortality of patients. So, early diagnosis and distinguishing malignancy from benign and premalignant lesions are particularly important. During the progression from benign neurofibromas to malignancy, a variety of changes including tumor morphology, genetic mutations, expression of multiple signaling pathways-related proteins and genome instability gradually occur. In this review, we detail these changes with the goals of identifying the histological and/or molecular signs of malignancy initiation, and an optimal therapeutic intervention window, to inhibit tumor progression and reduce the rate of mortality.
Cellular OncologyBiochemistry, Genetics and Molecular Biology-Cancer Research
CiteScore
10.40
自引率
1.50%
发文量
0
审稿时长
16 weeks
期刊介绍:
The Official Journal of the International Society for Cellular Oncology
Focuses on translational research
Addresses the conversion of cell biology to clinical applications
Cellular Oncology publishes scientific contributions from various biomedical and clinical disciplines involved in basic and translational cancer research on the cell and tissue level, technical and bioinformatics developments in this area, and clinical applications. This includes a variety of fields like genome technology, micro-arrays and other high-throughput techniques, genomic instability, SNP, DNA methylation, signaling pathways, DNA organization, (sub)microscopic imaging, proteomics, bioinformatics, functional effects of genomics, drug design and development, molecular diagnostics and targeted cancer therapies, genotype-phenotype interactions.
A major goal is to translate the latest developments in these fields from the research laboratory into routine patient management. To this end Cellular Oncology forms a platform of scientific information exchange between molecular biologists and geneticists, technical developers, pathologists, (medical) oncologists and other clinicians involved in the management of cancer patients.
In vitro studies are preferentially supported by validations in tumor tissue with clinicopathological associations.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


