Mayara Teixeira Alexandrino Sales, Rebeca Costa Castelo Branco, Carlos Henrique Paiva Granjeiro, Milena Silva Sousa, Luciana Felipe Férrer Aragão, Annelise Barreto de Carvalho, Ana Paula Dias Rangel Montenegro, Ana Rosa Pinto Quidute
{"title":"Multiple endocrine neoplasia type 1 in childhood and description of a novel variant.","authors":"Mayara Teixeira Alexandrino Sales, Rebeca Costa Castelo Branco, Carlos Henrique Paiva Granjeiro, Milena Silva Sousa, Luciana Felipe Férrer Aragão, Annelise Barreto de Carvalho, Ana Paula Dias Rangel Montenegro, Ana Rosa Pinto Quidute","doi":"10.1590/1984-0462/2025/43/2024175","DOIUrl":null,"url":null,"abstract":"<p><strong>Objective: </strong>To describe a case of multiple endocrine neoplasia type 1 in the pediatric age group and its molecular diagnosis.</p><p><strong>Case description: </strong>An 11-year-old boy began to present generalized tonic-clonic seizures in the presence of hypoglycemia, with high insulin dosage, leading to suspicion of insulinoma. Abdominal magnetic resonance imaging confirmed a pancreatic nodule, which was surgically resected, resulting in glycemic normalization. Low growth hormone levels and hyperprolactinemia, secondary to macroprolactinoma, were also identified. Treatment with cabergoline led to a reduction in size. Hyperparathyroidism was found asymptomatically, with parathyroid scintigraphy suggestive of adenoma, thus, the patient underwent subtotal parathyroidectomy and thymectomy with resolution of the condition. He entered puberty spontaneously at 15 years of age; however, he had decreased growth speed, short stature, and low insulin-like growth factor 1 (IGF-1) levels, indicating recombinant growth hormone. The next-generation sequencing panel for multiple endocrine neoplasia type 1 identified a probably pathogenic variant c.442A>C: p.(Thr148Pro) in heterozygosity in the MEN1 gene, without previous description in databases (ClinVar).</p><p><strong>Comments: </strong>We highlight the pre-pubertal age of multiple endocrine neoplasia type 1 diagnosis, which is made before age 21 in only 12-17% of cases, and hypoglycemia secondary to insulinoma as the initial manifestation, differing from what is most frequently described, namely prolactinoma and parathyroid adenoma. The clinical diagnosis was made based on the occurrence of two primary endocrine tumors and confirmed through a next-generation sequencing panel, with a variant not previously described in ClinVar.</p>","PeriodicalId":74721,"journal":{"name":"Revista paulista de pediatria : orgao oficial da Sociedade de Pediatria de Sao Paulo","volume":"43 ","pages":"e2024175"},"PeriodicalIF":2.0000,"publicationDate":"2025-03-24","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11940707/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Revista paulista de pediatria : orgao oficial da Sociedade de Pediatria de Sao Paulo","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1590/1984-0462/2025/43/2024175","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
Objective: To describe a case of multiple endocrine neoplasia type 1 in the pediatric age group and its molecular diagnosis.
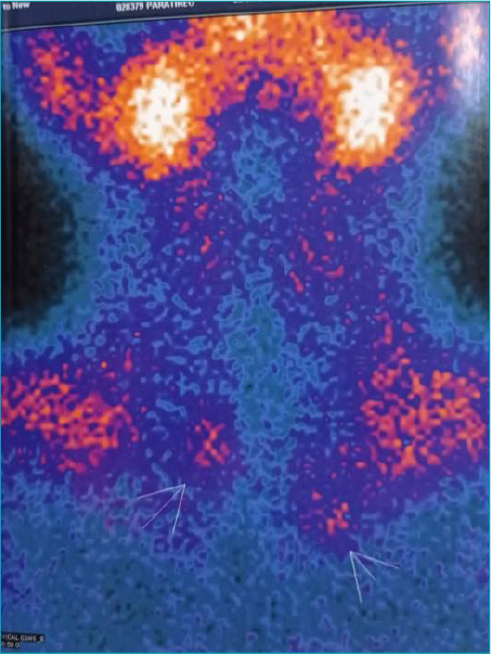
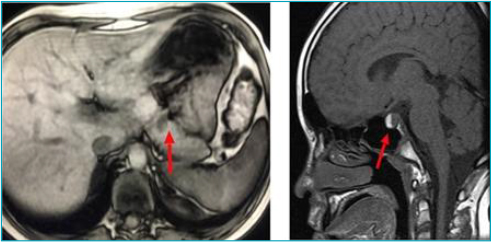
Case description: An 11-year-old boy began to present generalized tonic-clonic seizures in the presence of hypoglycemia, with high insulin dosage, leading to suspicion of insulinoma. Abdominal magnetic resonance imaging confirmed a pancreatic nodule, which was surgically resected, resulting in glycemic normalization. Low growth hormone levels and hyperprolactinemia, secondary to macroprolactinoma, were also identified. Treatment with cabergoline led to a reduction in size. Hyperparathyroidism was found asymptomatically, with parathyroid scintigraphy suggestive of adenoma, thus, the patient underwent subtotal parathyroidectomy and thymectomy with resolution of the condition. He entered puberty spontaneously at 15 years of age; however, he had decreased growth speed, short stature, and low insulin-like growth factor 1 (IGF-1) levels, indicating recombinant growth hormone. The next-generation sequencing panel for multiple endocrine neoplasia type 1 identified a probably pathogenic variant c.442A>C: p.(Thr148Pro) in heterozygosity in the MEN1 gene, without previous description in databases (ClinVar).
Comments: We highlight the pre-pubertal age of multiple endocrine neoplasia type 1 diagnosis, which is made before age 21 in only 12-17% of cases, and hypoglycemia secondary to insulinoma as the initial manifestation, differing from what is most frequently described, namely prolactinoma and parathyroid adenoma. The clinical diagnosis was made based on the occurrence of two primary endocrine tumors and confirmed through a next-generation sequencing panel, with a variant not previously described in ClinVar.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


